In this review article, we describe the diagnostic criteria (including the clinical and histological features) for immunoglobin G4-related kidney disease (IgG4-RKD), including the renal and other organ manifestations in IgG4-RKD, the pathophysiology and treatment.
Diagnostic Criteria
The diagnostic algorithm for IgG4-RKD includes the presence of kidney injury, as manifested by abnormal urinalysis or urine markers, abnormal radiologic findings or decreased kidney function with characteristic serological findings.6
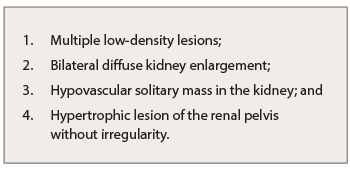
Table 1: Abnormal Renal Radiologic Findings of IgG4-RKD
Abnormal renal radiologic findings are usually the first distinctive elements of this rare entity (see Table 1). Of these, the most common radiologic features include multiple low-density lesions on enhanced computed tomography (CT). Bilateral diffuse kidney enlargement can appear in plain CT. In addition, radiology might detect a (rare) hypovascular solitary mass in the kidney—in that case, malignancy must be ruled out. The fourth radiologic abnormality includes hypertrophic lesion of the renal pelvis without irregularity of the renal pelvic surface. Urinary tract carcinoma is considered the most important condition in the differential diagnosis.7
Kidney injury is recognized by the presence of proteinuria, hematuria and elevated N-acetyl-β-D-glucosaminidase, β2-microglobulin and/or α1-microglobulin excretions in urinalysis (UA). Hypergammaglobulinemia or elevated serum IgG levels (>135 mg/dL), low complement and elevated serum IgE levels are all frequent serologic findings of IgG4-RKD, and at least one of these three abnormalities is necessary for the diagnosis (see Table 2 ).2,7

Table 2: Abnormal Laboratory Findings of IgG4-RKD
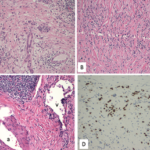
IgG4-related TIN (IgG4-RTIN) can be diagnosed using the organ-specific diagnostic criteria for IgG4-RKD. Characteristic histopathological findings of IgG4-RD include dense lymphoplasmacytic infiltration, which must be accompanied by >10 IgG4+ plasma cells/high power field (HPF) and/or IgG4+/IgG+ plasma cells >40%, and characteristic storiform fibrosis surrounding nests of lymphocytes and/or plasma cells (see Table 3). Therefore, lymphoplasmacytic TIN with sclerofibrosis and prominent IgG4-positive plasma cells appears to be a representative histological finding of IgG4-RKD.7,8 Other features useful for establishing a diagnosis include obliterative phlebitis, lesions extending into the renal capsule, eosinophil infiltration, well-defined regional lesion distribution and marked fibrosis (see Table 3). The histological findings in extra-renal organs include dense lymphoplasmacytic infiltration by >10 IgG4+ plasma cells/HPF, and/or IgG4+/IgG+ plasma cells >40%.8
Kawano et al. proposed diagnostic criteria for IgG4-RKD based on previously established diagnostic criteria for autoimmune pancreatitis (AIP).9 A diagnosis of IgG4-RKD is definitive in patients with organ enlargement, mass or nodular lesions, and/or organ dysfunction, serum IgG4 concentration >135 mg/dL and histopathological findings of >10 IgG4 cells/HPF and/or an IgG4+/IgG+ cell ratio >40% or characteristic fibrosis (see Table 4A). The diagnosis is probable in the presence of abnormal urinalysis or decreased kidney function with either elevated serum IgG4 levels, low complements or elevated serum IgE levels, and histological findings of >10 IgG4 cells/HPF and/or IgG4+/IgG+ cell ratio >40% or characteristic fibrosis. IgG4-RKD is also likely when abnormal renal radiologic findings are present, along with characteristic histologic findings in the kidney or extra-renal organs.
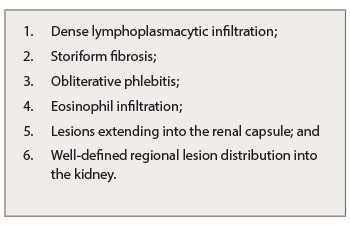
Table 3: Characteristic Histopathological Findings of IgG4-RKD
Finally, the diagnosis is probable when elevated serum IgG4 levels and histologic findings in the kidney are present (see Table 4B, p. 18). The diagnosis is possible when the presence of some kidney damage or abnormal renal radiologic findings are combined with either elevated serum IgG4 level or histological findings of >10 IgG4 cells/HPF and/or IgG4+/IgG+ cell ratio >40% (see Table 4C).10,11
Kidney Manifestations of IgG-RKD
Isolated kidney IgG4 disease has been reported and presents as multiple low-attenuation cortical nodules, wedge-shape lesions or diffuse patchy areas that may be associated with impaired renal function.10 The renal manifestation of IgG4-RD is, in most cases, tubulointerstitial, but sometimes may associate glomerular involvement, especially membranous nephropathy (MN), which could be also found as an isolated lesion (see Table 5).11
IgG4-RTIN can be focal or diffuse, and sometimes presents with tumor-like masses.12 Cystic dilatation of ducts can occur secondary to obstruction or stenosis of collecting ducts due to storiform or bird’s eye fibrosis.13 Obliterative phlebitis is an uncommon finding in kidney biopsy specimens because medium-size veins are rarely sampled.14 Only one case of IgG4-related renal arteritis has been described (see Table 5).15 Electron-dense deposits can be seen involving the tubular basement membrane.16 Immune complex deposits consist predominantly of IgG, C3, kappa and lambda light chains on immunofluorescence.17 In most cases, focal mild mononuclear cell tubilitis occurs, but eosinophilic or plasma cell tubulitis may also be seen.
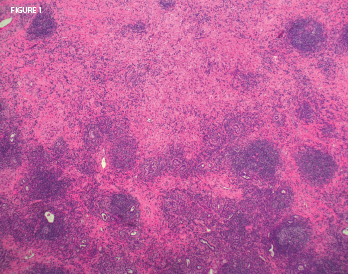
Figure 1: On biopsy, the lung mass lesion showed nodular inflammatory aggregates with residual alveoli and extensive lung fibrosis. Histopathologic image courtesy Dr. Paul Cohen, Yale University.
IgG4-RTIN can be present simultaneously in many cases of IgG4-related membranous glomerulonephritis (IgG4-RMGN) as detected by renal biopsy or suggested by kidney imaging findings (see Table 5). Approximately 7% of TIN cases include concurrent IgG4-RMGN.18 In some cases, glomerular lesions (i.e., IgA vasculitis, IgA nephritis, endocapillary proliferative glomerulonephritis, mesangial proliferative glomerulonephritis and membranoproliferative glomerulonephritis) have been associated with IgG4-RTIN.19 In membranous nephropathy (MN) associated with IgG4-RD, deposition of IgG4 in the glomerular basement membrane (GBM) has been demonstrated, either predominantly or along with other IgG subclasses. Although idiopathic MN and MN associated with IgG4-RD share a common immunologic process known as Th2 cytokine-mediated immune reaction and subsequent increase of IgG4, those two processes differ in the presence of the anti-PLA2R antibody.20 It has been reported in 70–80% of patients with idiopathic MN, and has been negative in all cases of IgG4-RD and also cases of MN associated with IgG4-RD, suggesting this antibody isn’t involved in the development of IgG4-RD.21
In idiopathic MN, the urine IgG4/IgG ratios were significantly higher in the idiopathic MN group, compared with the minimal-change disease or the focal segmental glomerulosclerosis group.22 Li et al. described a case of IgG4-related MN with high blood and low IgG4/IgG renal clearance ratio. Because IgG4 can engage the Fc of all IgG subclasses with its own Fc, potential binding between IgG4 and other molecules (IgG4 or other subclasses) might occur, and this might increase the molecule size and retard the renal filtration of IgG4. This observation could be a new clue to the different pathogenesis between IgG4-related MN and idiopathic MN, implying that charge selectivity barrier impairment might not occur in the pathogenesis of IgG4-related MN.23
Membranous glomerulonephritis (MGN) may be primary—in that case, it’s called idiopathic—or secondary. The majority of patients with primary MGN have antibodies against the M-type PLA2R antigen.24 MGN secondary to IgG4-RD can be called IgG4-related MGN, although primary MGN is also an IgG4-dominant disease.25 Therefore, immunostaining for IgG subclasses wouldn’t distinguish between primary MGN and IgG4-related MGN. However, staining for PLA2R is positive in the GBMs in the case of primary MGN, with a sensitivity of 74%.26

Table 4: Diagnostic Criteria for IgG4-RKD
Morimoto et al. described a rare case of membranoproliferative glomerulonephritis-like glomerular disease together with tubulointerstitial nephritis in association with AIP, as an organ expression of IgG4-RD.19 Other patterns of renal involvement include glomerular disease, ureteral inflammatory pseudotumor or retroperitoneal fibrosis (RF), which can cause renal failure and chronic sclerosing pyelitis (see Table 5, bottom right).27,28
In RF, the fibrotic lesion is macroscopically a firm white plaque around the abdominal aorta, illiacal vessels and ureters, whereas at the microscopic level, proliferation of fibroblasts and deposition of extracellular matrix may be noted. This fibro-inflammatory process results to hyalinization of the collagen.29
Other Organ Manifestations
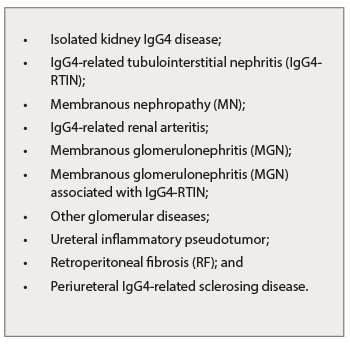
Table 5: Kidney Manifestations
of IgG4-RKD
Isolated IgG4-RKD remains uncommon.45 Most patients suffer multiorgan involvement. The most common extra-renal lesions include AIP, sialadenitis and dacryoadenitis (see Table 6).30
Sarles et al. first described AIP as a “chronic inflammatory sclerosis of the pancreas” in 1961.31 The different diagnostic criteria for AIP include imaging, laboratory, histopathology findings and response to steroid therapy. The Japan Pancreas Society (JPS) criteria, the Kim criteria, the HISORt criteria and the Italian criteria are used worldwide.32,33 The JPS, Kim and HISORt criteria are image based and include diffuse narrowing of the main pancreatic duct and diffuse enlargement of the pancreas with delayed (rim) enhancement. The laboratory criteria include elevated levels of serum gammaglobulins, elevated serum IgG4 and the presence of autoantibodies.34 All worldwide criteria employ the histopathology criteria, the response to steroid therapy and the association with either other organ involvement (biliary strictures, parotid/lacrimal gland involvement, mediastinal lymphadenopathy and RF) or other autoimmune diseases (such as ulcerative colitis, Crohn’s disease, sclerosing cholangitis, primary biliary cirrhosis and Sjӧgren’s syndrome [SS]). The histopathology criteria demonstrate marked lymphoplasmocytic sclerosing pancreatitis or lymphoplasmocytic infiltrate with dense fibrosis in the pancreas, or dense lymphocytic infiltration and destruction of the exocrine glands on histology.32,33,34
Hamano et al. reported that a level of IgG4 of 135 mg/dL and higher could differentiate AIP from pancreatic cancer, with a sensitivity of 95% and a specificity of 97%.28 The clinical manifestations can vary from a mild, acute recurrent pancreatitis, abdominal pain, anorexia and weight loss, to painless obstructive jaundice due to biliary stricture or a pancreatic mass.35

Table 6: Other Organ Manifestations of IgG-RKD
Mikulicz’s disease (MD), originally reported by Johann von Mikulicz-Radecki in 1892, is characterized by symmetric and persistent swelling of the lacrimal and salivary glands (see Table 6). MD has been recognized as a subtype of SS, which is a chronic form of dacryoadenitis and sialoadenitis. Mikulicz’s-type dacryoadenitis and sialoadenitis is associated with an elevated serum IgG4 level and fibrosis with significant infiltration of IgG4-positive plasma cells.36 Mavragani et al. found raised IgG4 serum levels in 7.5% of primary SS patients in association with IgG4-RD clinical, serologic and histopathologic features.37
Frequent complications of MD include AIP, retroperitoneal fibrosis and TIN with extraglandular involvement. Therefore, systemic examination and a fluorodeoxyglucose-positron emission tomography (FDG-PET) scan should be performed because multiple organ involvement might be present at initial diagnosis (see Fig. 1, and Fig. 2).38
First-line treatment is steroids with rapid disease response, but frequent flares can occur following discontinuation of glucocorticoid treatment.
Disease Pathophysiology
The presence of IgG4-positive plasma cells in the affected tissue and the fact that B cell depletion with rituximab (RTX) is effective in treatment suggest the importance of B-lymphocytes in the pathogenesis of IgG4-RD.39,40 Plasmablasts defined by cell surface expression of CD19, CD27 and CD38 (but negative for CD20) are elevated in the peripheral blood of patients with IgG4-RD correlating with disease activity. Because these plasmablasts demonstrate oligoclonal expansion and express IgG4, a specific antigen-driven immune response might be present in IgG4-RD.41,42
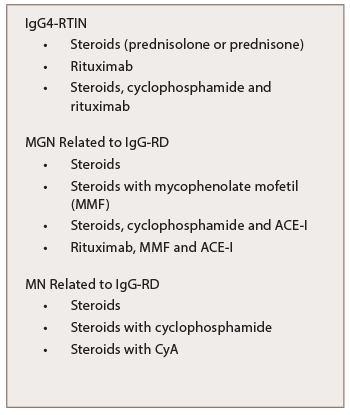
Table 7: Treatment of IgG-RKD
A Type 2 helper T cell (Th2)-driven immunological mechanism has been suggested in the pathogenesis of IgG4-RD as well.43 A clonally-expanded population of CD4+ cytotoxic T lymphocytes in the peripheral blood and fibrotic lesions of IgG4-RD patients suggest these cells are important in the disease pathogenesis by activating B cells.44 The Th2 cytokine IL-13 and the T-regulatory-associated cytokine transforming growth factor beta (TGF-β) are thought to activate fibroblasts and cause fibrosis, while the cytokines IL-4 and IL-10 might promote class-switching of IgG antibodies to IgG4 and differentiation of B cells into plasma cells. Polarized T cells possibly drive the storiform fibrosis and obliterative phlebitis observed in IgG4-RD. A separate T follicular helper cell response might generate the IgG4 phenotype.45
Memory CD4+ T cells produced by the continuous antigen presentation of B cells might explain the clinical improvement in B cell depletion. A parallel T follicular helper cell response, activated either by the same antigen or an event triggered by fibrosis, might induce the production of germinal centers within lymph nodes and generate plasmablasts that secrete IgG4. The latest studies have shown M2 macrophages activated by the cytokines such as IL-4 or IL-13 may also take part in the fibrosing process. Toll-like receptors on macrophages participate in class-switch recombination through B cell activating factor and could lead to further activation and proliferation of IgG4-positive B cells.46
Treatment
The first-line treatment is prednisolone at 0.6 mg/kg/day, or 30–40 mg/day of prednisolone as the initial dose to induce remission in type 1 AIP. Continue this dose for two to four weeks, and then gradually taper by 5 mg every one to two weeks to a maintenance dose of 5–10 mg/day.
Another approach: Taper 60 mg of prednisone over two to three months to a daily dose equivalent of between 2.5 and 10.0 mg/d of prednisone and then maintain this dose for several years.47 IgG4-TIN, even in cases with severe interstitial fibrosis, shows a brisk response to steroids (see Table 7). Although IgG4-RD shows a rapid response to steroids, the disease may recur after steroid tapering or withdrawal.48
In a cohort of 14 patients with no renal impairment (eGFR >60 mL/min/1.73 m2) but with imaging abnormalities, treatment with glucocorticoids improved the imaging abnormalities without a change in eGFR after a mean follow-up period of 42.4 ±17.0 months. Data from the same cohort indicated that 20% of patients treated with glucocorticoids experienced a relapse while on maintenance therapy (prednisolone median 5 mg daily), with a mean follow-up period of only 44 months.49 The natural course of IgG4-related TIN without treatment remains unknown. If a nontreatment strategy is chosen, close observation of renal function is warranted. Renal atrophy may occur in many patients with IgG-4-related TIN, and thus, treatment without overt clinical renal impairment is reasonable.
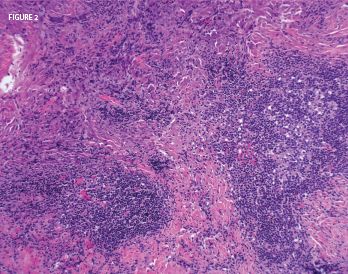
Figure 2: A nasal biopsy of a mass lesion showed lymphoid aggregates (abundant plasma cells) and dense fibrosis. Histopathologic image courtesy of Dr. Paul Cohen, Yale University.
In a study conducted by Quattrocchio et al., one patient with IgG4-RTIN was treated with steroids and rituximab, and four patients (two with IgG4-RTIN and two with IgG4-related RF) were treated with the following immune suppressive protocol for severe systemic lupus erythematosus nephritis: three pulses of 15 mg/kg intravenous methylprednisolone followed by oral tapering of prednisone plus two pulses of 500 mg intravenous cyclophosphamide (on days 1 and 15), plus four weekly rituximab administrations at 375 mg/m2. Two more doses of rituximab were administered one and two months after the last weekly infusion. Renal function improvement was observed in all patients (see Table 7).50
Experience with treating MGN associated with IgG4-RD was derived from a retrospective analysis of seven patients with a mean follow-up period of 39 months (ranging from 4–184 months). Four of those patients had concomitant biopsy proven IgG4-related TIN. Treatment of those seven patients varied: Two patients received prednisone; two initially were treated with prednisone alone and subsequently received prednisone with MMF; one received prednisone, cyclophosphamide and an angiotensin-converting-enzyme inhibitor (ACE-I); one received an ACE-I followed by rituximab and mycophenolate mofetil (MMF); and one patient went untreated (see Table 7). Data revealed a decline in proteinuria from a mean baseline of 8.8 g daily (3.5–16 g daily) to 1.2 g daily (ranging from 0–3.1 g daily) and an improvement in the mean serum creatinine concentration from 221.0 μmol/L (ranging from 70.7–583.4 μmol/L) to 123.8 μmol/L (ranging from 70.7–583.4 μmol/L).5
Kanda et al. treated a patient who developed repeated relapses of membranous nephropathy related to IgG4-RD initially with prednisolone, but after failure of this approach, treatment cyclophosphamide was added. Due to side effects of cyclophosphamide, the combination of prednisolone and cyclosporine A (CyA) was effective for the induction and maintenance of remission (see Table 7).51
In the event IgG4-RD is refractory or recurrent, the addition of immune-suppressants, such as azathioprine, methotrexate and MMF or rituximab, is recommended. Some patients may relapse after being treated with low doses of azathioprine (50 mg/d) or mycophenolate (1 g/d), but may respond to higher doses of these drugs.52 Rituximab is used to maintain disease remission with glucocorticoid treatment, and successful use of rituximab without associated glucocorticoids has been reported.53 B cell depletion not only interferes with antigen presentation by plasmablasts, it also reduces immune complex formation. The potential significance of immune complex formation as a possible disease mechanism seems relevant for patients with low complement levels— because IgG4 doesn’t bind complement well—and the manifestation of IgG-related TIN.54
In active IgG4-RD, circulating plasmablasts are elevated, so they could be used as a potential biomarker not only for diagnosis, but also for assessment of treatment response. Plasmablasts are circulating plasma cells that arise from activated CD20+ B cells and secrete antibodies. Anti-CD20 monoclonal antibodies function via antibody-dependent cell-mediated cytotoxicity and deplete circulating and tissue resident B cells, thus eliminating the progenitor to the plasmablast. Plasmablasts fall quickly after B cell depletion. The decline or recurrent rise of those cells sometimes correlates better with disease activity than the serum IgG concentration.42
Plasmablast-targeted therapy may offer a more specific treatment approach for IgG4-RD. XmAb5871, a monoclonal antibody with a high-affinity variable region binding to CD19 and an enhanced Fc domain that binds to the FcγRIIb inhibitory receptor of B cells, is currently in phase II development for IgG4-RD treatment and has already been studied in phase I trials in rheumatoid arthritis and systemic lupus erythematosus.55
Oligoclonally-expanded CD4+ effector-memory T cells with a cytotolytic phenotype (CD4+ CTLs) have been characterized recently in IgG4-RD. Those cells express SLAMF7, IL-1β, TGF-β1, granzyme B and perforin, and are the dominant T cells in the affected tissues. After rituximab administration, the concentrations and percentages of these novel T cells with CD4+CTLs phenotype decreased significantly but slower than the concentrations of B cells and plasmablasts. The responsiveness of those novel therapies to CD20-targeted B cell depletion is most likely attributed to the interference of T and B cells, because plasmablasts present antigen and activate memory CD4+CTLs at the inflammation site.56
Immunostaining for IgG subclasses wouldn’t distinguish between primary MGN & IgG4-related MGN. However, staining for PLA2R is positive in the GBMs in the case of primary MGN with a sensitivity of 74%.
Conclusions
One of the rare systemic manifestations of IgG4-RD is the kidney. IgG4-RTIN with characteristic imaging findings remains the most common type of kidney involvement in IgG4-RD. Glomerular diseases can develop simultaneously, or as isolated lesions in IgG4-RKD. The most common extra-renal manifestations include AIP, sialadenitis and dacryoadenitis. IgG4 is an important molecule, but its direct role in the disease pathogenesis remains unclear. On the other hand, plasmablasts are potential biomarkers of the disease, because they are found to be elevated in the peripheral blood of patients with IgG4-RD, correlating with disease activity and assisting with the optimal timing of treatment.
First-line treatment is glucocorticoids, which induce a rapid disease response, but frequent flares can occur following discontinuation of glucocorticoid treatment. In cases of recurrent disease or disease refractory to steroids the addition of immunosuppressants, such as rituximab, azathioprine, methotrexate, mycophenolate mofetil or cyclophosphamide, is recommended. Although CD20-targeted B cell depletion is a promising treatment, plasmablast-targeted treatment with monoclonal antibodies and novel therapies against CD4+ CTLs may be more specific and effective future treatment approaches for IgG4-RD. R
 Despina Michailidou, MD, PhD, is a first-year rheumatology fellow at the National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, Md. She obtained her MD from the National and Kapodistrian University of Athens in Greece. She received her PhD from the Goethe University of Frankfurt in Germany. She completed her residency in internal medicine at Yale New Haven Health System.
Despina Michailidou, MD, PhD, is a first-year rheumatology fellow at the National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, Md. She obtained her MD from the National and Kapodistrian University of Athens in Greece. She received her PhD from the Goethe University of Frankfurt in Germany. She completed her residency in internal medicine at Yale New Haven Health System.
 Paul J. Cohen, MD, chairs the Bridgeport Hospital Department of Pathology and Laboratory Medicine in Bridgeport, Conn. He spends most of his time in general surgical pathology and has a specialty interest in cardiovascular pathology. He’s an assistant professor of pathology at Yale School of Medicine in New Haven, Conn. He’s also the director of the Yale Pathology residency program at Bridgeport Hospital.
Paul J. Cohen, MD, chairs the Bridgeport Hospital Department of Pathology and Laboratory Medicine in Bridgeport, Conn. He spends most of his time in general surgical pathology and has a specialty interest in cardiovascular pathology. He’s an assistant professor of pathology at Yale School of Medicine in New Haven, Conn. He’s also the director of the Yale Pathology residency program at Bridgeport Hospital.
Acknowledgment: This review was supported in part by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health.
References
- Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012 Feb 9;366(6):539–551.
- Umehara H, Okazaki K, Masaki Y, et al. A novel clinical entity, IgG4-related disease (IgG4RD): General concept and details. Mod Rheumatol. 2012 Feb;22(1):1–14.
- Saeki T, Nishi S, Imai N, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int. 2010 Nov;78(10):1016–1023.
- Deshpande V, Chicano S, Finkelberg D, et al. Autoimmune pancreatitis: A systemic immune complex mediated disease. Am J Surg Pathol. 2006 Dec;30(12):1537–1545.
- Alexander MP, Larsen CP, Gibson IW, et al. Membranous glomerulonephritis is a manifestation of IgG4-related disease. Kidney Int. 2013 Mar;83(3):455–462.
- Umehara H, Okazaki K, Masaki Y, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012 Feb;22(1):21–30.
- Kawano M, Saeki T, Nakashima H, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011 Oct;15(5):615–626.
- Yamaguchi Y, Kanetsuna Y, Honda K, et al. Characteristic tubulointerstitial nephritis in IgG4-related disease. Hum Pathol. 2012 Apr;43(4):536–549.
- Okazaki K, Kawa S, Kamisawa T, et al. Clinical diagnostic criteria of autoimmune pancreatitis: Revised proposal. J Gastroenterol. 2006 Jul;41(7):626–631.
- Shoji S, Nakano M, Usui Y. IgG4-related inflammatory pseudotumor of the kidney. Int J Urol. 2010 Apr;17(4):389–390.
- Khosroshahi A, Stone JH. A clinical overview of IgG4-related systemic disease. Curr Opin Rheumatol. 2011 Jan;23(1):57–66.
- Cornell LD, Chicano SL, Deshpande V, et al. Pseudotumors due to IgG4 immune-complex tubulointerstitial nephritis associated with autoimmune pancreatocentric disease. Am J Surg Pathol. 2007 Oct;31(10):1586–1597.
- Fukuhara H, Taniguchi Y, Matsumoto M, et al. IgG4-related tubulointerstitial nephritis accompanied with cystic formation. BMC Urol. 2014 Jul 20;14:54.
- Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012 Sep;25(9):1181–1192.
- 15. Sharma SG, Vlase HL, D’Agati VD. IgG4-related tubulointerstitial nephritis with plasma cell-rich renal arteritis. Am J Kidney Dis. 2013 Apr;61(4):638–643.
- Tsubata Y, Akiyama F, Oya T, et al. IgG4-related chronic tubulointerstitial nephritis without autoimmune pancreatitis and the time course of renal function. Intern Med. 2010;49(15):1593–1598.
- Saeki T, Kawano M. IgG4-related kidney disease. Kidney Int. 2014 Feb;85(2):251–257.
- Stone JH. IgG4: A tantalizing link between causes of membranous glomerulonephritis and systemic disease. Kidney Int. 2013 Mar;83(3):348–350.
- Morimoto J, Hasegawa Y, Fukushima H, et al. Membranoproliferative glomerulonephritis-like glomerular disease and concurrent tubulointerstitial nephritis complicating IgG4-related autoimmune pancreatitis. Intern Med. 2009;48(3):157–162.
- Cravedi P, Abbate M, Gagliardini E, et al. Membranous nephropathy associated with IgG4-related disease. Am J Kidney Dis. 2011 Aug;58(2):272–275.
- Fervenza FC, Downer G, Beck LH Jr., et al. IgG4-related tubulointerstitial nephritis with membranous nephropathy. Am J Kidney Dis. 2011 Aug;58(2):320–324.
- Li J, Qu Z, Zhang YM, et al. [Clinical significance of detection of plasma and urine IgG4 in idiopathic membranous nephropathy]. Beijing Da Xue Bao Yi Xue Ban. 2010 Dec 18;42(6):671–674.
- Li XL, Yan TK, Li HF, et al. IgG4-related membranous nephropathy with high blood and low urine IgG4/IgG ratio: A case report and review of the literature. Clin Rheumatol. 2014 Jan;33(1):145–148.
- Beck LH Jr., Bonegio RG, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009 Jul;361(1):11–21.
- Imai H, Hamai K, Komatsuda A, et al. IgG subclasses in patients with membranoproliferative glomerulonephritis, membranous nephropathy and lupus nephritis. Kidney Int. 1997 Jan;51(1):270–276.
- Debiec H, Ronco P. PLA2R autoantibodies and PLA2R glomerular deposits in membranous nephropathy. N Engl J Med. 2011 Feb 17;364(7):689–690.
- Hamano H, Kawa S, Ochi Y, et al. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet. 2002 Apr 20;359(9315):1403–1404.
- Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. New Engl J Med. 2001 Mar 8;344(10):732–738.
- Van Moerkercke W, Verhamme M, Meeus G, et al. A case of IgG4-related sclerosing disease with retroperitoneal fibrosis, autoimmune pancreatitis and bilateral focal nephritis. Pancreas. 2009 Oct;38(7):825–832.
- Cornell LD. IgG4-related kidney disease. Curr Opin Nephrol Hypertens. 2012 May;21(3):279–288.
- Sarles H, Sarles JC, Camatte R, et al. Observations on 205 confirmed cases of acute pancreatitis, recurring pancreatitis and chronic pancreatitis. Gut. 1965 Dec;6(6):545–559.
- Kim KP, Kim MH, Kim JC, et al. Diagnostic criteria for autoimmune chronic pancreatitis revisited. World J Gastroenterol. 2006 Apr 28;12(16):2487–2496.
- Kwon S, Kim MH, Choi EK. The diagnostic criteria for autoimmune chronic pancreatitis: It is time to make a consensus. Pancreas. 2007 Apr;34(3):279–286.
- Chari ST, Smyrk TC, Levy MJ, et al. Diagnosis of autoimmune pancreatitis: The Mayo Clinic experience. Clin Gastroenterol Hepatol. 2006 Aug;4(8):1010–1016.
- Gardner TB, Chari ST. Autoimmune pancreatitis. Gastroenterol Clin North Am. 2008 Jun;37(2):439–460.
- Yamamoto M, Harada S, Ohara M, et al. Clinical and pathological differences between Mikulicz’s disease and Sjögren’s syndrome. Rheumatology (Oxford). 2005 Feb;44(2):227–234.
- Mavragani CP, Fragoulis GE, Rontogianni D, et al. Elevated IgG4 serum levels among primary Sjögren’s syndrome patients: Do they unmask underlying IgG4-related disease? Arthritis Care Res (Hoboken). 2014 May;66(5):773–777.
- Suga K, Kawakami Y, Hiyama A, et al. F-18 FDG PET-CT findings in Mikulicz disease and systemic involvement of IgG4-related lesions. Clin Nucl Med. 2009 Mar;34(3):164–167.
- Umehara H, Nakajima A, Nakamura T, et al. IgG4-related disease and its pathogenesis-cross-talk between innate and acquired immunity. Int Immunol. 2014 Nov;26(11):585–595.
- Khosroshahi A, Bloch DB, Deshpande V, et al. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010 Jun;62(6):1755–1762.
- Mattoo H, Mahajan VS, Della-Torre E, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol. 2014 Sep;134(3):679–687.
- Wallace ZS, Mattoo H, Carruthers M, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015 Jan;74(1):190–195.
- Lighaam LC, Aalberse RC, Rispens T. IgG4-related fibrotic diseases from an immunological perspective: Regulators out of control? Int J Rheumatol. 2012;2012:789164.
- Mattoo H, Mahajan VS, Maehara T, et al. Clonal expansion of CD4(+) cytotoxic T lymphocytes in patients with IgG4-related disease. J Allergy Clin Immunol. 2016 Sep;138(3):825–838.
- Yamamoto M, Takahashi H, Shinomura Y. Mechanisms and assessment of IgG4-related disease: Lessons for the rheumatologist. Nat Rev Rheumatol. 2014 Mar;10(3):148–159.
- Watanabe T, Yamashita K, Fujikawa S, et al. Involvement of activation of toll-like receptors and nucleotide-binding oligomerization domain-like receptors in enhanced IgG4 responses in autoimmune pancreatitis. Arthritis Rheum. 2012 Mar;64(3):914–924.
- Brito-Zeron P, Kostov B, Bosch X, et al. Therapeutic approach to IgG4-related disease: A systematic review. Medicine (Baltimore). 2016 Jun;95(26):e4002.
- Khosroshahi A, Stone JH. Treatment approaches to IgG4-related systemic disease. Curr Opin Rheumatol. 2011 Jan;23(1):67–71.
- Saeki T, Kawano M, Mizushima I, et al. The clinical course of patients with IgG4-related kidney disease. Kidney Int. 2013 Oct;84(4):826–833.
- Quattrocchio G, Roccatello D. IgG4-related nephropathy. J Nephrol. 2016 Aug;29(4):487–493.
- Kanda H, Koya J, Uozaki H, et al. Membranous nephropathy with repeated flares in IgG4-related disease. Clin Kidney J. 2013 Apr;6(2):204–207.
- Ghazale A, Chari ST, Zhang L, et al. Immunoglobulin G4-associated cholangitis: Clinical profile and response to therapy. Gastroenterology. 2008 Mar;134(3):706–715.
- Ebbo M, Daniel L, Pavic M, et al. IgG4-related systemic disease: Features and treatment response in a French cohort: Results of a multicenter registry. Medicine (Baltimore). 2012 Jan;91(1):49–56.
- Kawano M, Mizushima I, Yamaguchi Y, et al. Immunohistochemical characteristics of IgG4-related tubulointerstitial nephritis: Detailed analysis of 20 Japanese cases. Int J Rheumatol. 2012;2012:609795.
- Chu SY, Yeter K, Kotha R, et al. Suppression of rheumatoid arthritis B cells by XmAb5871, an anti-CD19 antibody that coengages B cell antigen receptor complex and Fcgamma receptor IIb inhibitory receptor. Arthritis Rheumatol. 2014 May;66(5):1153–1164.
- Perugino CA, Stone JH. Treatment of IgG4-related disease: Current and future approaches. Z Rheumatol. 2016 Sep;75(7):681–686.