
When elevated, the enzyme most considered to be a sign of myositis is creatine phosphokinase (CPK). It is sometimes mistakenly believed that CPK must be elevated at least one time during the course of polymyositis, but this is not always the case. Some patients have normal CPK levels throughout their course. Furthermore, there are several other causes of an elevated CPK, including exercise (anaerobic and aerobic), trauma (blunt or sharp), drugs (statins, colchicine, alcohol, cocaine), and toxins. Carriers for muscular dystrophy or some metabolic myopathies may have an increased serum CPK. Healthy African-American males commonly have CPK levels above the upper limits of normal reported for the population as a whole. Finally, in large clinical trials using statin drugs, over 30% of individuals have elevated CPK levels. Interestingly, the percentage was the same for those taking placebo as it was for those taking study drugs.3 Thus, relying on this test as a way to screen for muscle inflammation can be problematic.
The classic triad of EMG changes described includes: 1) fibrillation at rest, increased insertional activity, and spontaneous and positive sharp waves; 2) bizarre high-frequency repetitive discharges; and 3) polyphasic potentials of short duration and low amplitude.1,2 However, this triad is not diagnostic and is generally present in only 40% of patients with myositis, while 10% of patients with these diseases have normal EMG results.4
Similar issues are observed when studying the histopathology of these diseases. For example, the finding of lymphocytes in and around muscle fibers or in a perivascular location is consistent with the diagnosis of polymyositis or dermatomyositis. Yet, it is not uncommon to see nonspecific myopathic changes or even normal histology on muscle biopsy in some patients suspected of having an inflammatory myopathy.
According to Bohan and Peter, four criteria are needed to confirm definite disease, three to confirm probable disease, and two to confirm possible disease.1,2 Though this approach sounds simplistic, it can be difficult to apply this standard to individual patients. For example, is it appropriate to prescribe high-dose corticosteroids or other immunosuppressive therapies for a patient who fulfills just two criteria?
Bohan and Peter subdivided inflammatory myopathies into a number of subtypes including primary idiopathic polymyositis, primary idiopathic dermatomyositis, dermatomyositis (or polymyositis) associated with neoplasia, childhood dermatomyositis (or polymyositis) associated with vasculitis, and polymyositis or dermatomyositis associated with collagen-vascular disease (overlap group).1,2 Inclusion body myositis, first described in 1979, has subsequently been added as another subtype. Once again, this categorization is probably too simplistic.
Today, we believe that patients with classic forms of polymyositis, dermatomyositis, and inclusion body myositis have very characteristic changes in muscle histology. For example, polymyositis is characterized by infiltration of CD8+ lymphocytes invading muscle fibers with degenerating and regenerating fibers. In contrast, dermatomyositis has CD4+ T lymphocytes, B cells, and plasmacytoid dendritic cells in perivascular locations and perifascicular atrophy. Inclusion body myositis is associated with changes identical to polymyositis, but also demonstrates lined vacuoles and deposits of beta-amyloid and ubiquitin. These changes suggest a different underlying pathophysiology for each of the three conditions. However, the variations in pathology (variations that overlap or fail to show all the features described above) that are seen in so many patients suggest there are far more than just these three phenotypes. For example, one patient may have the pathology described for polymyositis, but also a rash characteristic of dermatomyositis. Another patient may show the reverse. Thus, it may be challenging to accurately diagnose some patients.
The identification of myositis-specific autoantibodies in many patients with inflammatory myopathies has increased our diagnostic abilities to some degree (see Table 2). Although a patient with the classic manifestation of the antisynthetase syndrome will have polymyositis or dermatomyositis plus interstitial lung disease, arthritis, fever, Raynaud’s phenomenon, and mechanic’s hands, there are other patients with circulating antisynthetase antibodies who have pulmonary disease without muscle involvement, or myositis without the other features. The presence of anti–signal recognition particle (SRP) antibodies usually carries a very poor prognosis, yet some patients with these antibodies respond well to therapy. Similarly, anti-Mi2 antibodies are generally, though not always, associated with a very good prognosis.

The Differential Diagnosis Is Huge
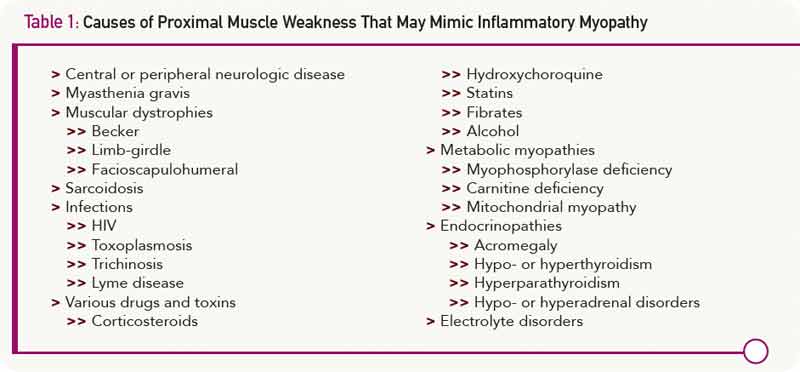
Another challenge in diagnosing idiopathic inflammatory myopathies involves differentiating them from many other conditions, considered to be myositis mimics, which can also fulfill Bohan and Peter’s criteria (see Table 1). Bohan and Peter recognized the relative nonspecific nature of their criteria, stating, “There are certain findings, herein called exclusions, whose presence should make one very cautious about rendering a diagnosis of polymyositis.”2
The first group of myositis mimics are muscular dystrophies. These are inherited disorders characterized by progressive muscle weakness that advances to muscle wasting. Becker, limb-girdle, and facioscapular dystrophies may present in late adolescence or adulthood with proximal muscle weakness and elevated muscle enzymes. Although the classic histologic picture in these diseases includes muscle-fiber size variation with necrosis and eventually replacement of muscle by fat and fibrous tissue, inflammatory cells may also be observed in early stages. The situation may be further complicated by the observation that some patients with muscular dystrophy note a temporary improvement with corticosteroid therapy.
Selected infections can cause a chronic myopathy. This is particularly true for immunocompromised individuals in whom toxoplasmosis infection can result in the development of an inflammatory myopathy. Some patients infected with HIV develop a slowly progressive proximal myopathy with elevated CPK levels. Muscle biopsy demonstrates endomysial, perimysial, or perivascular mononuclear cell infiltrates and muscle fiber necrosis.
Many drugs can induce a myopathy. Corticosteroids and HMG-CoA reductase inhibitors (statins) are among the most common. Corticosteroid use can result in the development of painless proximal muscle weakness, especially involving the pelvic girdle muscles and neck flexors. In patients being treated for diseases other than myositis, the serum CPK is normal and magnetic resonance imaging of the muscles is unremarkable. EMG may show low-amplitude motor unit potentials, and muscle biopsy classically demonstrates type 2 fiber atrophy. The clinical picture may be confounding when a myositis patient treated with corticosteroids initially improves to a degree and then begins to regress. Is the myositis flaring, or is there a superimposed corticosteroid myopathy? Generally, the latter condition occurs in patients who have been treated with corticosteroids for at least four weeks or, in most cases, for much longer. It is more likely to occur in patients treated with higher doses of corticosteroids, especially the fluorinated preparations (triamcinolone, dexamethasone, or betamethasone). One strategy to diagnose steroid myopathy is to rapidly taper the dose and see if there is clinical improvement or worsening.
More than 10% of patients treated with statins will complain of muscle problems. Symptoms include myalgia, cramping, tenderness, or weakness often occurring in pectoral, quadriceps, biceps, abdominal, and low back muscles. CPK values range from normal to more than 10 times the upper limit of normal. Some patients develop an apparent immune-mediated necrotizing myopathy.5 This may be associated with circulating antibodies to HMG-CoA reductase and respond to vigorous immunosuppressive therapy.
Other drugs can also induce a polymyositis-like picture. Examples include penicillamine, interferon-α, and procainamide. Hydroxychloroquine, drugs that cause hypokalemia (diuretics and amphotericin), and licorice-induced hypokalemia can cause proximal muscle weakness. A common histopathologic feature for all these drugs is the presence of vacuoles seen on muscle biopsy.
Among the metabolic myopathies, myophosphorylase deficiency (McArdle disease), carnitine deficiency, and some mitochondrial myopathies can cause proximal muscle weakness, elevated CPK levels, and myopathic EMG changes. Muscle biopsies are not uniformly helpful since they may show nonspecific changes. The diagnosis may require sophisticated metabolic testing.
Evidence Regarding Therapy Is Lacking
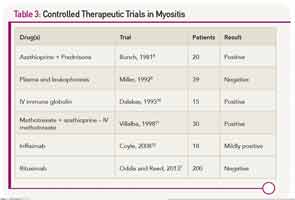
There are scant evidenced-based data to refer to when making therapeutic decisions for myositis (see Table 3). This may be one of the reasons why the prognosis for patients with polymyositis and dermatomyositis has not changed since the introduction of corticosteroids in 1949. About 20% to 40% of treated patients can enter a period of sustained remission, with the remaining 60% to 80% experiencing a continuous or fluctuating course.6 A concomitant malignancy, or the presence of circulating anti-SRP antibodies, suggest a poor prognosis. In most other situations, one cannot readily predict which patients will be good or poor responders.
Because idiopathic inflammatory myopathies are rare diseases, often have a very insidious onset (especially for inclusion body myositis), and can be challenging to diagnose, there have been few sizeable clinical trials. All but one of the published controlled trials had fewer than 50 patients. That study, the Rituximab in Myositis (RIM) trial, enrolled more than 200 patients from more than 30 centers. Despite the fact that more than 80% of subjects met the definition of improvement, the primary endpoints were not met.7
In the future, therapy may be guided by genetic or molecular testing. For example, rituximab may be more effective in patients with selected genetic signatures. Similarly, anti–interferon-α antibody (a biologic agent under development) may be effective in patients whose muscle has evidence for increased levels of interferon-α inducible genes. In the meantime, therapy for polymyositis and dermatomyositis remains empiric.
Dr. Jones is assistant professor of medicine and Dr. Wortmann is professor emeritus of medicine at the Geisel School of Medicine at Dartmouth-Hitchcock Medical Center in Lebanon, N.H. Disclosure: Dr. Wortmann is on an advisory board for Questcor Pharmaceuticals.
References
- Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
- Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975;292:403-407.
- Law M, Rudnicka AR. Statin safety: A systematic review. Am J Cardiol. 2006;97(8A):52C-60C.
- Sultan SM, Ioannou Y, Moss K, Isenberg DA. Outcome in patients with idiopathic inflammatory myositis: Morbidity and mortality. Rheumatology (Oxford). 2002;41:22-26.
- Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. 2010;62:2757-2766.
- Marie I. Morbidity and mortality in adult polymyositis and dermatomyositis. Curr Rheumatol Rep. 2012;14:275-285.
- Oddis CV, Reed AM, Aggarwal R, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: A randomized, placebo-phase trial. Arthritis Rheum. 2013;65:314-324.
- Bunch TW. Prednisone and azathioprine for polymyositis: Long-term followup. Arthritis Rheum. 1981;24:45-48.
- Miller FW, Leitman SF, Cronin ME, et al. Controlled trial of plasma exchange and leukapheresis in polymyositis and dermatomyositis. N Engl J Med. 1992;326:1380-1384.
- Dalakas MC, Illa I, Dambrosia JM, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329:1993-2000.
- Villalba L, Hicks JE, Adams EM, et al. Treatment of refractory myositis: A randomized crossover study of two new cytotoxic regimens. Arthritis Rheum. 1998;41:392-399.
- Coyle K, Pokrovnichka A, French K, et al. A randomized, double-blind, placebo-controlled trial of infliximab in patients with polymyositis and dermatomyositis. Arthritis Rheum. 2008;58(Suppl):Abstract: 2058.

