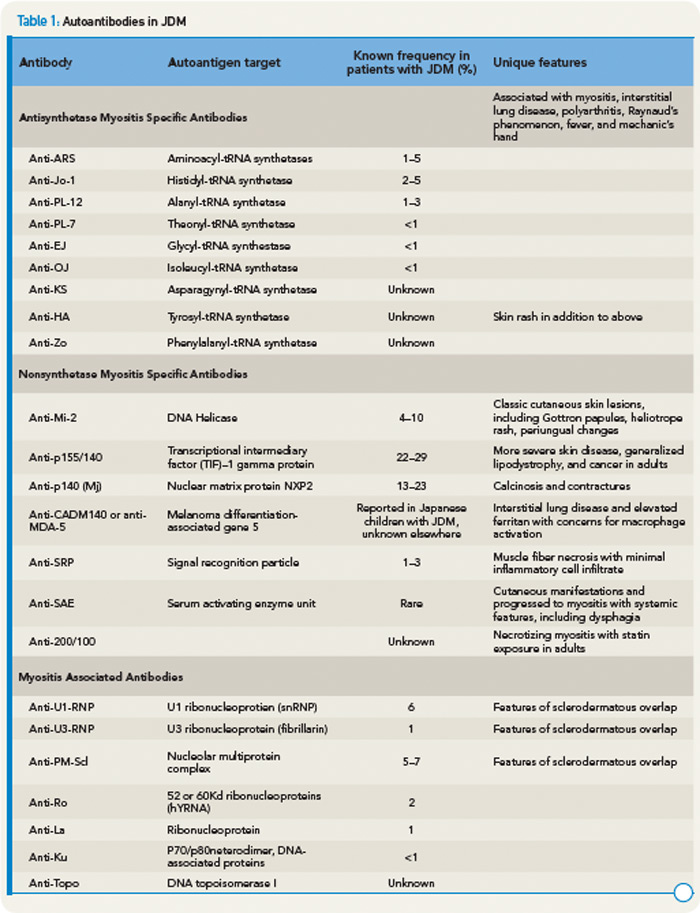
Recently, anti-p155/140 antibodies have been reported in 23% to 29% of JDM, and anti-p140 autoantibodies alone are seen in 20% to 30% of JDM and are associated with increased risk of calcinosis.12,13 Another 140kDa autoantibody, anti-MJ, is associated with severe disease with contractures and atrophy.14 While MSAs and MAAs are not always performed in children, they may be beneficial to help with prognosis and atypical clinical findings.
Recent Research Findings
It has long been appreciated that both humoral (autoantibodies and immune complexes) and cellular (T and B cells) components of the adaptive immune system contribute to the pathogenesis of JDM. However, in the last several years, there has been increasing evidence that the innate immune system, especially the upregulation of type I interferons and plasmacytoid dendritic cells may play an important role in the pathogenesis of disease.


