Thus, this child has classic findings of JDM—what would be initial therapy options?
JDM is the most common subset of the juvenile idiopathic inflammatory myopathies (IIM), representing about 85% of the IIM cases in patients aged 16 years or younger.1 The incidence of JDM in the United States is 3.2 per million children per year.2 It is more common in females, with an average age of onset of seven years of age. One-fourth of the patients are less than four years of age at onset.3
Clinical Features of JDM
JDM usually presents as easy fatigability, irritability in a young child, and inability or refusal to perform normal daily activity such as getting up off the floor, climbing stairs, playing, dressing, etc. The onset of muscle weakness is usually insidious and, at times, blamed on behavior issues. There is usually difficulty with and gradually increasing fatigue in activities of daily living, such as combing hair, reaching for highly placed objects, climbing stairs, or getting in or out of bed. The onset of rash, muscle weakness, and fatigability may be months apart or in the same time frame.
The skin rash is variable, yet usually includes erythematous lesions on extensor surfaces especially in sun-exposed areas such as the small joints of the hands (Gottron’s papules), knees, elbows, and medial malleoli of the ankles. The classic heliotrope rash of the eyelids, periorbital edema, and erythematous malar rash that can occur in a mask-like distribution is often confused as a systemic lupus erythematosus rash. Psoriasiform lesions can occur on the extensor surfaces and scalp, with alopecia in a smaller subset of patients. Nailfold and gingival telangiectasias are seen commonly and may result in bleeding of the gums. Cutaneous ulcerations, when present, are thought to represent a severe phenotype of the disease. An amyopathic form of JDM occurs in <5% of children.4 Calcinosis is seen in 20% to 30% of children with JDM and may present as small punctate lesions, especially in areas of trauma (elbows, knees, waist), tumoral, sheet-like masses, or liquefied collections.5
Other clinical findings seen with JDM include fever, weight loss, shortness of breath, dysphagia, dysphonia (such as nasal or hoarse voice), Raynaud’s phenomenon, and arthritis. In severe disease, dysphagia, cutaneous ulcers, gastrointestinal ulcers, or features of a vasculopathy may all require immediate immunosuppression. Pulmonary dysfunction is also common and mainly due to weakness in the respiratory musculature. Interstitial lung disease can also be seen in JDM, though less frequently than in adults.6
Cardiac involvement can lead to conduction abnormalities and pericarditis/myocarditis. Other less frequent clinical findings include poikilodermatous skin lesions, lipodystrophy, retinitis, iritis, seizures, and renal impairment.
There is symmetric proximal muscle weakness with and without elevated levels of serum muscle enzymes including creatine kinase (CK), aldolase, aspartate transaminase (AST), alanine transaminase, and lactate dehydrogenase (LDH). When elevated, these results help to differentiate disease quiescence or remission from active disease. However, a subset of subjects may have normal values at diagnosis and during times of muscle disease activity. CK blood levels may have a poor correlation with disease activity because CK levels become less sensitive with a greater duration of the disease.7 CK and the other enzymes may be discordant with severity of muscle disease on biopsy and with clinical muscle strength improvement. The level of muscle enzyme elevation early in disease may not predict long-term outcomes. Some clinicians believe that LDH and AST in combination may be the best indicators of disease flare.
Lymphocyte counts and subset characterization, neopterin, and von Willebrand factor products may serve as sensitive markers in a subset of patients.8,9
Autoantibody Profile in JDM
ANA is present in more than 70% of children.10 Rheumatoid factor and antibodies to SSA, SSB, Sm, RNP, and DNA may rarely be seen in some patients. Antibodies to PM/SCL identify a small, distinct subset of patients with a protracted disease course, often complicated by pulmonary interstitial fibrosis and/or cardiac involvement as well as sclerodactyly.14
The presence of myositis-specific autoantibodies (MSAs) is rare in children. Anti-Jo-1 antibodies are the most prevalent, found in about 2% to 5% of JDM patients, and anti-Mi-2 antibody is seen in 5% (see Table 1).11 Antibodies targeting signal recognition particles (SRP) are seen in 2% to 3% of JDM. They share the same clinical presentation as adults with an acute onset of severe myositis of both proximal and distal muscles, myocarditis, and a resultant severe disability.
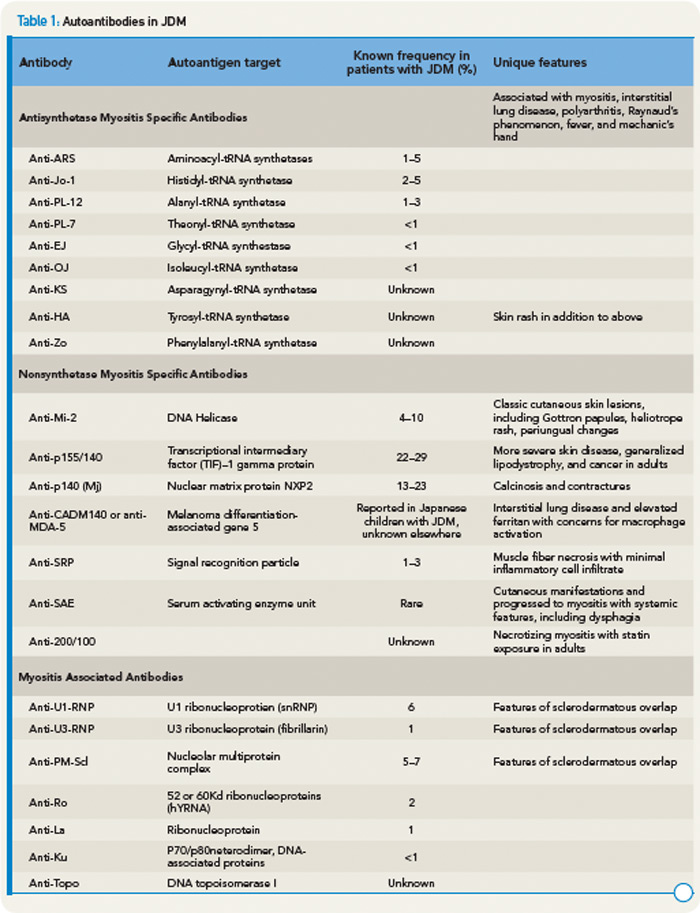
Recently, anti-p155/140 antibodies have been reported in 23% to 29% of JDM, and anti-p140 autoantibodies alone are seen in 20% to 30% of JDM and are associated with increased risk of calcinosis.12,13 Another 140kDa autoantibody, anti-MJ, is associated with severe disease with contractures and atrophy.14 While MSAs and MAAs are not always performed in children, they may be beneficial to help with prognosis and atypical clinical findings.
Recent Research Findings
It has long been appreciated that both humoral (autoantibodies and immune complexes) and cellular (T and B cells) components of the adaptive immune system contribute to the pathogenesis of JDM. However, in the last several years, there has been increasing evidence that the innate immune system, especially the upregulation of type I interferons and plasmacytoid dendritic cells may play an important role in the pathogenesis of disease.
Gene expression profiling of peripheral blood and affected tissues in JDM has revealed important insights into the molecular pathways underlying autoimmunity. The most prominent and consistent finding of these studies has been the presence of a gene signature characteristic of type I interferon (IFN) pathway activation, discovered first in DM muscle tissue and later identified in peripheral blood cells.15 The type I IFN pathway was further identified to include the cytokines/chemokines (MxA, IP-10, ITAC, MCP, interleukin 6) related to upregulation of this pathway.19
What do these studies tell us about disease mechanisms in JDM? Like many autoimmune diseases, there seems to be a genetic as well as environmental component. The genetic component appears to be multifactorial; HLA class genotype, especially HLA DQA1*0501 may help with persistence or development of maternal microchimerism.16 Cytokine polymorphisms may be involved as well. Whether there is an environmental trigger is unknown. Once the disease is initiated, there is maturation of dendritic cells leading to a type 1 interferon response and a cycle of inflammation develops with resulting damage to muscle and vascular endothelium, activation of B cells and CD4+ T cells, and overexpression of MHC class 1 on cell surfaces. It is believed that once this cycle begins, it may be halted by immunosuppressive therapy.
Therapy and Future Directions
The mortality rate of JDM was markedly reduced following the inception of corticosteroid therapy. With the use of methotrexate, the mean time of corticosteroid use fell to 10 months, compared with a historical mean time of 27 months.17 Surveys of North American pediatric rheumatologists indicate that most practitioners will treat typical JDM with corticosteroids and methotrexate, although there is great discrepancy between dosing regimens.18 Medications for refractory disease vary, but include hydroxychloroquine, intravenous immunoglobulin, rituximab, cyclophosphamide, cyclosporine A, azathioprine, tacrolimus, and mycophenolate mofetil.
There is no specific evidence basis for the treatment of calcinosis. Anti–tumor necrosis factor agents, bisphosphonates, and calcium channel blockers have been used. Surgical excision may be indicated in areas of recurrent infection, chronic pain, or impedance of function due to calcinosis.
The treatment of resistant skin disease is controversial—topical corticosteroids and pimecrolimus may help with symptomatic itching or redness, but it is possible that resistant skin disease reflects ongoing systemic disease and should be treated with increasing immunosuppression.
Given the findings of increased serum CD19+ B cells and lymphoid follicles in muscle biopsies of affected patients, rituximab has been proposed as a possible therapy. Initial case series noted clinical improvements in skin and muscle that paralleled B-cell depletion.19 A large, randomized, controlled, crossover trial of rituximab in refractory JDM and adult DM/polymyositis (the Rituximab in Myositis Study) was devised where patients refractory to corticosteroids and at least one other immunosuppressive agent were given rituximab or placebo on Weeks 0, 1, 8, and 9. The authors found that 83% of patients with refractory adult and juvenile myositis met the definition of improvement, but there was no significant difference in time to improvement.20 It was suggested that there may be better response to rituximab in the juvenile cohort within this study.
The Childhood Arthritis and Rheumatology Research Alliance (CARRA) has developed consensus-driven treatment protocols for patients with moderately severe JDM. These include combinations of oral corticosteroids, pulse intravenous corticosteroids, methotrexate, and intravenous immunoglobulin.21 Use of consensus protocols over time will hopefully lead to a more evidence-based rationale for treatment in the future.
Conclusion
While the causes of juvenile and adult DM are still elusive, recent developments point to the involvement of interferon and lymphoid follicles in the pathogenesis of DM. An initial aggressive approach using corticosteroids and disease-modifying antirheumatic drugs such as methotrexate may result in better long-term outcomes with less calcinosis and corticosteroid toxicity. In children, the prognosis is generally favorable, although future goals should include minimizing treatment toxicities and long-term morbidities of disease (calcinosis, lipodystrophy, cardiac, and pulmonary complications). Identifying biomarkers that can predict disease outcome and prognosis will be a challenge for dermatomyositis researchers in the future.
Disclosures
Dr. Reed has been a consultant for Genentech. Dr. Robinson has no disclosures.
Dr. Robinson is assistant professor of pediatric rheumatology at Rainbow Babies and Children’s Hospital in Cleveland. Dr. Reed is professor of pediatrics and medicine in the department of pediatric rheumatology at Mayo Clinic in Rochester, Minn.
References
- Rider LG, Miller FW. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies. JAMA. 2011;305:183-190.
- Mendez EP, Lipton R, Ramsey-Goldman R, et al. US incidence of juvenile dermatomyositis, 1995-1998: Results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheum. 2003;49:300-305.
- Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008;371:2201-2212.
- Klein RQ, Teal V, Taylor L, Troxel AB, Werth VP. Number, characteristics, and classification of patients with dermatomyositis seen by dermatology and rheumatology departments at a large tertiary medical center. J Am Acad Dermatol. 2007;57:937-943.
- Eidelman N, Boyde A, Bushby AJ, et al. Microstructure and mineral composition of dystrophic calcification associated with the idiopathic inflammatory myopathies. Arthritis Res Ther. 2009;11:R159.
- Sanner H, Aalokken TM, Gran JT, Sjaastad I, Johansen B, Flato B. Pulmonary outcome in juvenile dermatomyositis: A case-control study. Ann Rheum Dis. 2011;70:86-91.
- Guzman J, Petty RE, Malleson PN. Monitoring disease activity in juvenile dermatomyositis: The role of von Willebrand factor and muscle enzymes. J Rheumatol. 1994;21:739-743.
- Robinson AB, Reed AM. Clinical features, pathogenesis and treatment of juvenile and adult dermatomyositis. Nature Rev Rheumatol. 2011;7:664-675.
- DeBenedetti F, DeAmici M, Aramini L, Ruperto N, Martini A. Correlation of serum neopterin concentrations with disease activity in juvenile dermatomyositis. Arch Dis Child. 1993;69:232-235.
- Wedderburn LR, McHugh NJ, Chinoy H, et al. HLA class II haplotype and autoantibody associations in children with juvenile dermatomyositis and juvenile dermatomyositis-scleroderma overlap. Rheumatology (Oxford). 2007;46:1786-1791.
- Wedderburn LR, Rider LG. Juvenile dermatomyositis: New developments in pathogenesis, assessment, and treatment. Best Pract Res Clin Rheumatol. 2009;23:665-678.
- Gunawardena H, Wedderburn LR, North J, et al. Clinical associations of autoantibodies to a p155/140 kDa doublet protein in juvenile dermatomyositis. Rheumatology. 2008;47:324-328.
- Gunawardena H, Wedderburn LR, Chinoy H, et al. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthritis Rheum. 2009;60:1807-1814.
- Espada G, Maldonado Cocco JA, Fertig N, Oddis CV. Clinical and serologic characterization of an Argentine pediatric myositis cohort: Identification of a novel autoantibody (anti-MJ) to a 142-kDa protein. J Rheumatol. 2009;36:2547-2551.
- Bilgic H, Ytterberg SR, Amin S, et al. Interleukin-6 and type I interferon-regulated genes and chemokines mark disease activity in dermatomyositis. Arthritis Rheum. 2009;60:3436-3446.
- Reed AM, Stirling JD. The HLA-DQA1*0501 allele in multiple racial groups with juvenile dermatomyositis. Hum Immunol. 1995;44:131-135.
- Ramanan AV, Campbell-Webster N, Ota S, et al. The effectiveness of treating juvenile dermatomyositis with methotrexate and aggressively tapered corticosteroids. Arthritis Rheum. 2005;52:3570-3578.
- Stringer E, Bohnsack J, Bowyer SL, et al. Treatment approaches to juvenile dermatomyositis (JDM) across North America: The Childhood Arthritis and Rheumatology Research Alliance (CARRA) JDM Treatment Survey. J Rheum. 2010;37:1953-1961.
- Rios Fernandez R, Callejas Rubio JL, Sanchez Cano D, Saez Moreno JA, Ortego Ceteno N. Rituximab in the treatment of dermatomyositis and other inflammatory myopathies. A report of 4 cases and review of the literature. Clin Exp Rheumatol. 2009;27:1009-1016.
- Oddis CV, Reed AM, Aggarwal R, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis (DM) and adult polymyositis (PM)—The RIM Study [abstract L13]. Arthritis Rheum. 2010;62:3844.
- Huber AM, Giannini EH, Bowyer SL, et al. Protocols for the initial treatment of moderately severe juvenile dermatomyositis: Results of a Children’s Arthritis and Rheumatology Research Alliance Consensus Conference. Arthritis Care Res. 2010;62:219-225.


