Since the introduction of angiotensin-converting enzyme inhibitors and their remarkable success in the management of scleroderma renal crisis, pulmonary complications have assumed ever greater importance as a source of morbidity and mortality in this disease.
Signs and Symptoms
Dyspnea is the most common complaint of patients with ILD or PAH. Like hunger or thirst, dyspnea can be characterized as a “synthetic sensation” in that it often arises from multiple sources of information rather than from stimulation of a single neural receptor. Severity of dyspnea as well as qualitative aspects of unpleasant breathing experiences vary widely among patients.1 Dyspnea usually signals the presence of lung or heart disease (or rarely, coexisting muscle disease). Standardized tools exist to quantify dyspnea, and these are often used in clinical trials; in the office it is more practical to assess dyspnea in terms of self-reported activities (e.g., walking, carrying, and climbing stairs).
Paul Klee, the famous 20th-century artist who suffered from scleroderma, described his symptoms while hiking in the mountains: “I inhale as deep as possible. My dyspnea depends on the trail, whether going up or going down. It also depends upon the weather … and it also depends on how much food I have in my stomach.”2 With progressive disease, even simple tasks may prove difficult. Near the end his life, Klee noted that the small incline to his house was difficult to traverse: “This is now my Matterhorn.”
Because the intensity of dyspnea associated with ambulatory activity depends on the rate of work performance (power output), patients may reduce the rate of work performance, thereby minimizing the intensity or distress of symptoms.1 Frequently, my patients tell me that they experience no breathlessness as long as they perform tasks slowly. Other patients find their activity limited by articular or neuromuscular complications—or simply by the overwhelming fatigue associated with SSc. The complaint of dyspnea is an important clue to the presence of underlying lung or cardiac disease; but the absence of dyspnea does not rule out significant pulmonary disease.
Usually, dyspnea associated with SSc is chronic. When a patient with this disease presents with acute dyspnea, the rheumatologist must consider pulmonary embolism, pneumothorax, infection, serositis, and heart failure. Acute pulmonary edema may be a manifestation of volume overload secondary to scleroderma renal crisis, other causes of renal failure, or primary scleroderma heart disease.
Cough is another symptom of lung fibrosis and usually is nonproductive and made worse by exertion. Productive cough or hemoptysis requires evaluation to rule out aspiration pneumonia or neoplasm—each of which occurs with increased frequency in SSc.
Physical examination remains key when evaluating any rheumatic disease patient. Some argue cynically that the stethoscope is obsolete in this era of high-tech diagnostic studies. To others, “the stethoscope embodies the essence of doctoring: using science and technology in concert with the human skill of listening to determine what ails a patient.”3 Laennec’s invention of the stethoscope initiated medicine’s irreversible trend toward physically separating the diagnosing physician from the patient.3 High-tech medicine has expanded the potential gap between patient and physician. For me, the hollow tube of the stethoscope is a connection to my patient and reinforces my need to listen.
Fine inspiratory (and sometimes expiratory) crackles are a finding heard in patients with ILD. Crackles may be audible at the lung bases and higher in more extensive disease; in early disease, however, crackles may not be audible. Accentuation of the pulmonic component of S2, a tricuspid regurgitation murmur, or right-sided S4 may be indicative of PAH. Right ventricular heave, jugular venous distension, and edema are later findings of PAH with right-sided heart failure. The presence of any of these physical findings requires further evaluation but, like dyspnea, their absence does not preclude ILD or PAH.
Pulmonary Function Testing
Perform pulmonary function tests (PFTs) at baseline and regular intervals in all patients. The characteristic PFT pattern of SSc-ILD is restriction, with reduced lung volumes (e.g., total lung capacity) and a proportionate reduction in diffusion capacity for carbon monoxide (DLCO). In the absence of significant obstructive airway disease, the forced vital capacity (FVC) serves as a surrogate for total lung capacity. If the FVC percent predicted is less than 75% and the ratio of the FVC percent predicted divided by the DLCO percent predicted is <1.4, the patient has a predominantly restrictive PFT pattern characteristic of ILD.
SSc patients with PAH have a disproportionate decline in DLCO relative to decline in FVC. A fall in DLCO may be the first clue to the presence of SSc-PAH, and, if confirmed by serial testing, work-up for PAH is warranted. If the FVC percent predicted is greater than 75% and the ratio of the FVC percent predicted divided by the DLCO percent predicted is >1.4, the patient has predominantly pulmonary vascular disease. Of course, mixed patterns may occur in patients with both ILD and PAH.
Six-Minute Walk Test
The six-minute walk test (6MWT) is a sub-maximal exercise test that yields important information about exercise capacity of the patient and oxygen saturation. (See Figure 1, page 18.) This test is useful to obtain at baseline and, when performed serially, can be used to monitor response to therapy. In some pulmonary conditions, the 6MWT correlates with New York Heart Association functional classification and even predicts mortality. Such correlations have not been demonstrated in SSc, perhaps due to extra-pulmonary features of SSc (e.g., joint pain, muscle weakness, etc.) that often affect ambulation. Nevertheless, the distance walked can be monitored and compared with prior test results for the individual patient. In addition, if exercise-induced hypoxemia is present, supplemental oxygen should be prescribed.
Imaging Studies
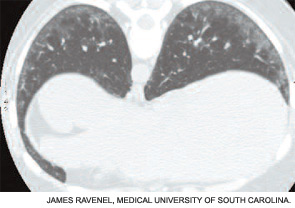
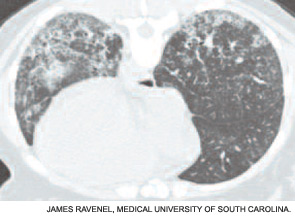
Although the chest radiograph is not a sensitive test for detecting early ILD, it may detect lung infiltrates or nodules, pleural disease, lymphadenopathy, or cardiomegaly. High-resolution computed tomography (CT) is a much more sensitive test and should be part of the baseline assessment of all SSc patients. The earliest sign of ILD is a hazy-appearing opacification through which normal lung architecture can be seen (ground-glass opacification). (See Figure 2A, page 18.) With progression, more advanced signs of disease (e.g., honeycombing and traction bronchiectasis) may be seen. (See Figure 2B, page 18.) Patients with a normal CT scan have a good long-term prognosis, whereas those with CT abnormalities tend to worsen if not treated.
Pulmonary thromboembolism rarely occurs in SSc but suspect it in any patient who experiences acute dyspnea or in the patient with risk factors for chronic thromboembolic disease (e.g., sedentary lifestyle or antiphospholipid antibody). Ventilation-perfusion lung scans and CT pulmonary angiography are indicated in such situations.

Bronchoalveolar Lavage
In years past, SSc-ILD was believed to result from a bland fibrosing process. My lab was among the first to employ bronchoscopy with lavage to obtain cells and proteins from the lower respiratory tract of SSc patients, and these studies provided new insight into the pathogenesis of SSc-ILD.5,6 Many patients have evidence of an inflammatory process characterized by increased numbers of neutrophils and eosinophils, activated alveolar macrophages, and increased levels of pro-inflammatory and pro-fibrotic cytokines. Cells cultured from bronchoalveolar lavage fluid exhibit the phenotype of an activated myofibroblast with contractile and synthetic properties.7 The origin of these cells is not certain, but these activated myofibroblasts are key players in the pathogenesis of SSc-ILD.
Although bronchoalveolar lavage remains a valuable research tool, it may be less useful in the clinic. For technical reasons, the interpretation of bronchoalveolar lavage fluid can be difficult when performed in community-based laboratories. Another disadvantage of this test is the potential for sampling error because generally only one or two sites are lavaged. When performed in a standardized fashion in specialized laboratories, however, it does provide clues to the presence of active ILD.

Lung Biopsy
Open lung biopsy is rarely indicated unless there is clinical concern for other pulmonary conditions that might confound SSc (e.g., bronchiolitis obliterans with organizing pneumonia, sarcoidosis, or aspiration, among others). The predominant histologic pattern of SSc-ILD is a nonspecific interstitial pneumonitis. CT findings in SSc closely resemble those observed in patients with idiopathic nonspecific interstitial pneumonitis.
Echocardiography
Obtain echocardiogram with Doppler at baseline on all newly diagnosed patients, and perform serial studies at intervals, depending upon the patient’s clinical symptoms and the results of their other tests (e.g., DLCO). Echocardiography is an important tool to evaluate the complaint of dyspnea, particularly when PAH is suspected (e.g., in the patient found to have a low DLCO percent predicted with normal or only slightly reduced FVC percent predicted [vide supra]).
In two recent studies conducted in community-based rheumatology practices, the prevalence of PAH as defined by an estimated RV systolic pressure of >40 mm Hg was found to be approximately 25% in SSc and mixed connective tissue disease patients.8,9 Thus, one must have a high index of suspicion for PAH in this patient population.
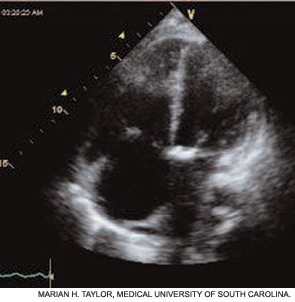
Echocardiography provides important information about right atrial (RA) and right ventricular (RV) size and function—especially RV systolic pressure. (See Figure 3, page 18.) Congenital heart disease, valvular disease, pericardial effusion, and left-sided heart dysfunction may also be detected.
Right Heart Catheterization
Because the Doppler echocardiogram may overestimate or underestimate RV systolic pressure, right heart catheterization is required whenever PAH is suspected. Right heart catheterization remains the gold standard for accurate diagnosis of PAH and also permits assessment of concomitant left ventricular disease by measuring pulmonary capillary wedge pressure or left ventricular end-diastolic pressure. PAH is defined as a mean pulmonary artery pressure of >25 mm Hg at rest, or >30 mm Hg with exercise, and a normal pulmonary capillary wedge pressure (<15 mm Hg). Diastolic dysfunction is not an uncommon cause for dyspnea in SSc patients. Treatment of diastolic dysfunction differs significantly from that of PAH; therefore, cardiac catheterization is critical for accurate diagnosis and management of the SSc patient with suspected pulmonary vascular disease.
Diagnosis and Management
My recommended baseline cardiopulmonary evaluation for a newly diagnosed SSc patient is shown in Table 2. (See above right.) If baseline studies are normal and the patient has no complaints of dyspnea, I recommend annual re-assessment (sooner if the patient develops symptoms). If PFTs are abnormal, further work-up is necessary and should be guided by the particular pattern of PFT abnormalities. For example, a restrictive PFT pattern suggests SSc-ILD, which should be confirmed by CT.
Other causes of restriction must be considered, especially if the CT is normal. If the PFT pattern suggests pulmonary vascular disease, then echocardiography is indicated. Obstructive airway disease is seen in some SSc patients—especially former or current smokers, those with asthma, chronic obstructive pulmonary disease, or bronchiolitis, or patients with secondary Sjogren syndrome. Manage dyspnea due to obstructive airway disease with bronchodilator drugs and measures to prevent gastroesophageal reflux disease (GERD).
If the echocardiogram reveals evidence of PAH (e.g., right atrial enlargement, RV hypertrophy, asymmetric septal bowing, or elevated RV systolic pressure), perform a right heart catheterization to rule out PAH. In some SSc patients, dyspnea is the result of diastolic dysfunction and heart failure; in such cases the echocardiogram may reveal left atrial enlargement, left ventricular hypertrophy, abnormal E/A slope, or findings of left-sided diastolic dysfunction. It is essential to distinguish left-sided heart failure from PAH, since the management of pulmonary venous hypertension differs greatly from that of PAH.
Log on to www.The-Rheumatologist.org for printable flowcharts on PFT and echocardiogram assessment.
Treatment of SSc-ILD
Once you establish a diagnosis of SSc-ILD, treatment is warranted—especially for those patients who have relatively early disease. The best and most current evidence for treatment of SSc-ILD derives from the Scleroderma Lung Study: a double-blind, randomized controlled trial to test the efficacy of cyclophosphamide for the treatment of SSc-ILD.10 Scleroderma Lung Study patients had dyspnea, restrictive lung physiology, and evidence of inflammatory ILD either by bronchoalveolar lavage, high-resolution computed tomography, or both. Treatment consisted of oral cyclophosphamide, <2 mg/kg daily or placebo for one year with an additional one year of follow-up. For the primary end-point, FVC percent predicted at 12 months, there was a clinically modest and statistically significant difference in favor of cyclophosphamide (2.53%, 95% CI 0.28% to 4.79%, p<0.03). The difference in FVC was maintained at 24 months.
Interestingly, a significant interaction between baseline fibrosis on CT and treatment was seen: patients in the placebo group with high baseline fibrosis scores had the greatest decline in FVC at one year, whereas baseline degree of fibrosis had no significant influence on the change in FVC for the cyclophosphamide treatment group. Also of interest, cyclophosphamide therapy was associated with statistically significant differences in dyspnea score, total lung capacity, skin score, and certain health-related measures of quality of life. Significantly more adverse effects were observed in the cyclophosphamide treatment group than in the placebo group, so treated patients must be monitored carefully for signs or symptoms of drug toxicity. Less robust studies suggest a similar beneficial effect on FVC with other immunosuppressive agents (e.g., azathioprine and mycophenolate mofetil), but randomized controlled trials are needed to test the efficacy of these agents for SSc-ILD. For those patients who fail all therapy, lung transplantation is an option but only at selected transplant centers.
Treatment of SSc-PAH
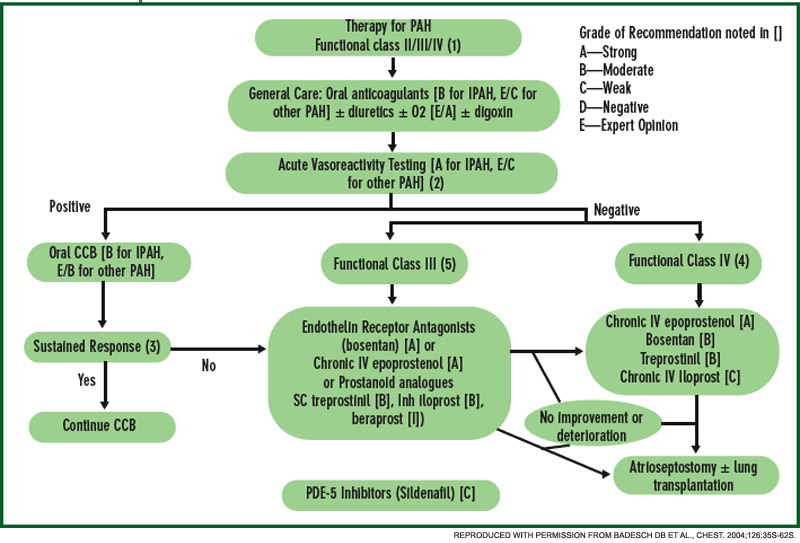
Current therapy is targeted to one or more of the major pathogenic mediators of PAH—endothelin-1, prostacyclin (PGI2), and nitric oxide. Evidence supports the use of agents that block endothelin-1 (endothelin receptor antagonists), augment PGI2 (PGI2 and its analogues), or enhance nitric oxide (phosphodiesterase-5 inhibitors). Guidelines for treatment of PAH have been developed by the
American College of Chest Physicians.11 (See Figure 4.) Several caveats apply to SSc-PAH. First, oral anticoagulation may not be feasible if GERD is severe. Also, SSc patients rarely display acute vasoreactivity and are not likely to experience improvement in PAH with calcium channel blocker therapy. Finally, SSc-PAH patients do not fare as well as patients with idiopathic PAH (previously known as primary pulmonary hypertension).
Bosentan, an oral, dual ETA/ETB receptor antagonist has been approved for the treatment of patients with class III or class IV PAH. It improves exercise capacity (6MWT) and hemodynamics, and reduces time to clinical worsening when used in doses of 125 mg bid after an initial month of treatment at 62.5 mg bid. Recent reports suggest that first-line bosentan therapy, with the subsequent addition of other PAH treatments if required, is safe and may prolong survival.12 Bosentan treatment can be associated with untoward effects—hepatotoxicity, anemia, teratogenecity, etc. Monitoring of liver tests is required prior to initiating bosentan therapy and monthly thereafter. Exclude pregnancy and encourage patients to prevent it by two forms of birth control.
Sildenafil is a phosphodiesterase-5 inhibitor approved for the treatment of patients with class II, III, or IV PAH. In doses of 20, 40, or 80 mg tid, sildenafil improves exercise capacity, hemodynamics, and functional class. More studies are needed before it can be determined if sildenafil improves time to clinical worsening or improves survival. The recommended starting dose is 20 mg tid. Sildenafil is well tolerated but may cause headache, flushing, dyspepsia, and epistaxis. A clinical trial is underway to assess the efficacy and safety of bosentan and sildenafil combination therapy.
PGI2 and its analogues are approved for the treatment of patients with class III or IV PAH. Epoprostenol is administered by continuous intravenous infusion, whereas trepostinil can be administered by subcutaneous or intravenous infusion. Sudden interruption in drug delivery may be complicated by extreme elevation of pulmonary artery pressure. Administer iloprost by inhalation six to nine times daily. Combination therapy with bosentan and inhaled iloprost improves exercise capacity and time to clinical worsening.
Conclusion
Great strides have been made in understanding and managing the major pulmonary complications of SSc, and the prospect of even better treatment has never been greater. Management of SSc lung disease requires the combined skills of the rheumatologist, pulmonologist, radiologist, and cardiologist. The rheumatologist plays a vital role in this process, first by exercising a high index of suspicion to ensure early diagnosis, and then by coordinating the complex management of the SSc patient. Rheumatologists are in the unique position of seeing patients prior to the development of end-stage ILD or PAH. It is our responsibility to see that SSc patients undergo full evaluation for what are now treatable complications of their disease.
Dr. Silver is professor of medicine and pediatrics at the Medical University of South Carolina in Charleston. He would like to thank his colleagues in the Pulmonary Hypertension Center at the Medical University of South Carolina (MUSC) and the patients and staff of the MUSC Scleroderma Clinic.
Michele Kaufman is a freelance medical writer based in New York City and a clinical pharmacist at New York Downtown Hospital.
References:
- Dyspnea. Mechanisms, assessments, and management: a consensus statement. Am J Respir Crit Care Med. 1999;159:321-340.
- Ostendorf B, Maiburg B, Schneider M. Scleroderma and Paul Klee: Metamorphosis of life and art (Title in German). Z Rheumatol. 2004;63:318-325.
- Markel H. The stethoscope and the art of listening. N Engl J Med. 2006;354:551-553.
- Launay D, Remy-Jardin M, Michon-Pasturel U, et al. High resolution computed tomography in fibrosing alveolitis associated with systemic sclerosis. J Rheumatol. 2006;33:1789-1801.
- Silver RM, Metcalf JF, Stanley JH, LeRoy EC. Interstitial lung disease in scleroderma. Analysis by bronchoalveolar lavage. Arthritis Rheum. 1984;27: 12541262.
- Ludwicka A, Trojanowska M, Smith EA, et al. Growth and characterization of fibroblasts obtained from bronchoalveolar lavage of scleroderma patients. J Rheumatol. 1992;19:1716-1723.
- Bogatkevich GS, Ludwicka-Bradley A, Nietert PJ, Silver RM. Scleroderma lung fibroblasts: Contractility and connective tissue growth factor. In: Tissue Repair, Contraction and the Myofibroblast, Desmouliere A, Gabbiani G, eds., Springer Science + Business Media, 2006.
- Wigley FM, Lima JA, Mayes M, et al. The prevalence of undiagnosed pulmonary arterial hypertension in subjects with connective tissue disease at the secondary health care level of community-based rheumatologists (the UNCOVER study). Arthritis Rheum. 2005;52:2125-2132.
- Pope JE, Lee P, Baron M, et al. Prevalence of elevated pulmonary arterial pressures measured by echocardiography in a multicenter study of patients with systemic sclerosis. J Rheumatol. 2005;32:1273-1278.
- Tashkin DP, Elashoff R, Clements PJ, et al. for the Scleroderma Lung Study. Cyclophosphamide versus placebo in scleroderma lung disease. N Eng J Med. 2006;354:2655-2666.
- Badesch DB, Abman SH, Ahearn GS, et al. Medical therapy for pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126:35S-62S.
- Denton CP, Humbert M, Rubin L, Black CM. Bosentan treatment for pulmonary arterial hypertension related to connective tissue disease: a subgroup analysis of the pivotal clinical trials and their open-label extensions. Ann Rheum Dis. 2006;65:1336-1340.
