Other causes of restriction must be considered, especially if the CT is normal. If the PFT pattern suggests pulmonary vascular disease, then echocardiography is indicated. Obstructive airway disease is seen in some SSc patients—especially former or current smokers, those with asthma, chronic obstructive pulmonary disease, or bronchiolitis, or patients with secondary Sjogren syndrome. Manage dyspnea due to obstructive airway disease with bronchodilator drugs and measures to prevent gastroesophageal reflux disease (GERD).
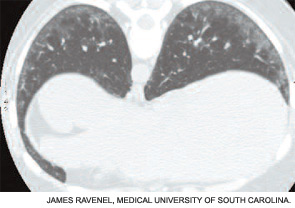
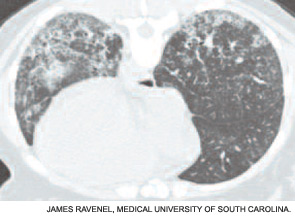
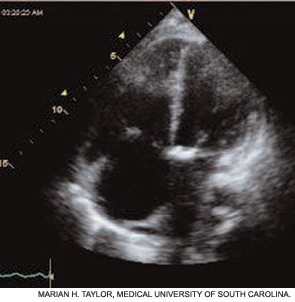
If the echocardiogram reveals evidence of PAH (e.g., right atrial enlargement, RV hypertrophy, asymmetric septal bowing, or elevated RV systolic pressure), perform a right heart catheterization to rule out PAH. In some SSc patients, dyspnea is the result of diastolic dysfunction and heart failure; in such cases the echocardiogram may reveal left atrial enlargement, left ventricular hypertrophy, abnormal E/A slope, or findings of left-sided diastolic dysfunction. It is essential to distinguish left-sided heart failure from PAH, since the management of pulmonary venous hypertension differs greatly from that of PAH.
Log on to www.The-Rheumatologist.org for printable flowcharts on PFT and echocardiogram assessment.
Treatment of SSc-ILD
Once you establish a diagnosis of SSc-ILD, treatment is warranted—especially for those patients who have relatively early disease. The best and most current evidence for treatment of SSc-ILD derives from the Scleroderma Lung Study: a double-blind, randomized controlled trial to test the efficacy of cyclophosphamide for the treatment of SSc-ILD.10 Scleroderma Lung Study patients had dyspnea, restrictive lung physiology, and evidence of inflammatory ILD either by bronchoalveolar lavage, high-resolution computed tomography, or both. Treatment consisted of oral cyclophosphamide, <2 mg/kg daily or placebo for one year with an additional one year of follow-up. For the primary end-point, FVC percent predicted at 12 months, there was a clinically modest and statistically significant difference in favor of cyclophosphamide (2.53%, 95% CI 0.28% to 4.79%, p<0.03). The difference in FVC was maintained at 24 months.
Interestingly, a significant interaction between baseline fibrosis on CT and treatment was seen: patients in the placebo group with high baseline fibrosis scores had the greatest decline in FVC at one year, whereas baseline degree of fibrosis had no significant influence on the change in FVC for the cyclophosphamide treatment group. Also of interest, cyclophosphamide therapy was associated with statistically significant differences in dyspnea score, total lung capacity, skin score, and certain health-related measures of quality of life. Significantly more adverse effects were observed in the cyclophosphamide treatment group than in the placebo group, so treated patients must be monitored carefully for signs or symptoms of drug toxicity. Less robust studies suggest a similar beneficial effect on FVC with other immunosuppressive agents (e.g., azathioprine and mycophenolate mofetil), but randomized controlled trials are needed to test the efficacy of these agents for SSc-ILD. For those patients who fail all therapy, lung transplantation is an option but only at selected transplant centers.
Treatment of SSc-PAH
Current therapy is targeted to one or more of the major pathogenic mediators of PAH—endothelin-1, prostacyclin (PGI2), and nitric oxide. Evidence supports the use of agents that block endothelin-1 (endothelin receptor antagonists), augment PGI2 (PGI2 and its analogues), or enhance nitric oxide (phosphodiesterase-5 inhibitors). Guidelines for treatment of PAH have been developed by the
American College of Chest Physicians.11 (See Figure 4.) Several caveats apply to SSc-PAH. First, oral anticoagulation may not be feasible if GERD is severe. Also, SSc patients rarely display acute vasoreactivity and are not likely to experience improvement in PAH with calcium channel blocker therapy. Finally, SSc-PAH patients do not fare as well as patients with idiopathic PAH (previously known as primary pulmonary hypertension).
