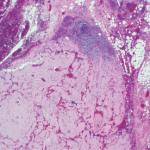
Yet the discovery of amyloid predates this astute characterization. In 1853, a decade before Dr. Alzheimer’s birth, another German professor and physician, Rudolf Virchow, MD, identified a form of amyloid when he examined deposits of this seemingly abnormal extracellular material in liver tissue and described how it reacted with iodine and sulfuric acid by turning blue, confirming the presence of starch. He coined the term “amyloid,” based on the Greek word amylon, meaning starch.4
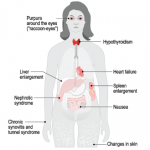
So what, after all, is amyloid? The amyloidoses comprise a group of protein-misfolding diseases that can complicate chronic inflammatory disorders, such as rheumatoid arthritis, or chronic infections (AA amyloidosis) or may develop in the setting of excess light chain production in patients with multiple myeloma (AL amyloidosis).
Amyloid fibrils are derived from the acute-phase reactant serum amyloid A (SAA) protein, an apolipoprotein constituent of high-density lipoprotein that is synthesized by hepatocytes under the transcriptional regulation of pro-inflammatory cytokines, particularly interleukin-6.2 The fibrils are composed of proteins that often have been mutated, partially fragmented or otherwise altered, which predisposes them to adopt an abnormal conformation.
Sustained overproduction of SAA is a prerequisite for the development of AA amyloidosis; although for reasons that are poorly understood, amyloidosis occurs only in a small proportion of patients with chronic inflammatory disorders. Interestingly, in Simone’s case, we never identified an underlying inflammatory disease.
The Amyloid Hypothesis
In the brain, amyloid precursor protein is cleaved to form amyloid beta (Aβ), a 40-amino-acid-long peptide that can aggregate in neurons to form soluble structures.5 However, when improperly folded, the tangled protein gets tossed into the cell, creating a bit of a mess, which in cell biology is never a good thing, because these structures steadily accumulate inside the cell. Lacking mechanisms for their proper disposal, the tangles become toxic to nerve cells. Over time, as more and more neurons become afflicted and then impaired, memory loss and personality changes ensue, leading to the clinical and pathological state so elegantly described by Dr. Alzheimer more than a century ago.
This concept of amyloid accumulation forms the basis for what is known as the amyloid hypothesis, the predominant framework for the pathogenesis of Alzheimer’s disease (AD). Aβ plaques are thought to wreak havoc by blocking proteasome function, inhibiting mitochondrial activity, altering intracellular Ca2+ levels and stimulating inflammatory processes.
Most importantly, Aβ plaques interact with the signaling pathways that regulate the phosphorylation of the microtubule-associated protein tau. Hyperphosphorylation of tau disrupts its normal function in regulating axonal transport and leads to the accumulation of neurofibrillary tangles and toxic species of soluble tau.5 In fact, accumulation of tau, rather than amyloid, may be the true culprit for the development of cognitive impairment.
The intense interest in the biology of amyloid has shifted from the body to the brain, where it continues to gain interest as a major target of study among neuroscientists.
Cognitively normal people can accumulate a large amyloid load in their brains, so the simple accumulation of amyloid in the brain is insufficient to cause dementia. However, there appears to be a much clearer correlation between the number of neurofibrillary tangles containing tau found post-mortem and the degree of dementia observed in life.6 Thus, a new view proposes that Aβ plaque formation is the key initiator of a complex pathogenic cascade that causes AD.