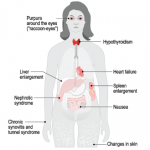
A cursory set of lab tests ordered in the emergency department provided a few clues: Anemia, elevated creatinine and frothy urine signified a heavy degree of protein loss. Unlike our current system, in which the average inpatient length of stay is often measured in hours, this was the predigital era, when the pace was slower and patients could spend days or weeks wandering and wondering on the ward as their doctors trudged steadily toward a diagnosis. I got to know Simone well as our team worked to untangle her disparate issues—the kidney disease, the constipation and the baffling observation that she had no arterial pulses to be felt anywhere.
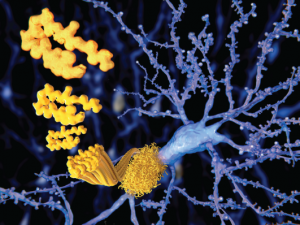
The beta amyloid peptid, amyloid plaques growing on a neuron.
Juan Gaertner / Shutterstock.com
We identified some interesting clues, including a thickened tongue that seemed to get in the way of her speaking fluently, a peripheral neuropathy that explained her reluctance to venture too far from her bed and many, many bruises that would spontaneously erupt over her waxy skin.
We were closing in on the diagnosis. We had a suspicion that an infiltrative disorder could explain her multisystem demise. A skin and fat pad biopsy was planned for the following day. But it never happened: That evening, Simone was found dead in her bed. A sudden death.
The pathologist who performed the autopsy was awestruck. In all his years of dissecting the bodies of the deceased in search of answers, he had never observed such a cause of death. His thick Glaswegian brogue was almost indecipherable to my Canadian ears, but the evidence that he held in his hands was irrefutable.
The normally flaccid bowel appeared rod-like rigid, looking and feeling more like a stovepipe, its walls studded with stercoral ulcers, those rarely seen perforations caused by chronic, massive fecal impaction explaining why Simone had been complaining so bitterly.1 Mon dieu! A picture is worth a thousand words. The pervasive infiltration of amyloid throughout her body had rendered her blood vessels pulseless and wrecked her kidneys, too.
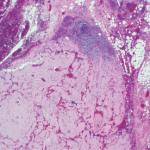
As we peered through microscopes to study these tissues, the striking apple green birefringence of the Congo red stain confirmed that their destruction had been caused by amyloid, the starch-like protein whose predilection to invade and damage selected tissues and organs is well known to rheumatologists.
Perhaps its penchant for linking to the glycosaminoglycans that form the backbone of connective tissue led to amyloid crossing the rheumatologist’s path periodically. Recall the well-described, though rarely seen, shoulder pad sign—a condition caused by the accumulation of massive sheets of amyloid material in the shoulder joints, giving the patient, usually a frail individual in their eighth decade of life or older, the appearance of a football player donning shoulder pads.2 There are also those rare occasions in which the ensuing synovial thickening of the knuckles that was characteristic of amyloid arthritis, misled clinicians into believing the patient had rheumatoid arthritis. These vignettes recall a bygone era when amyloidosis seemed more prevalent.
From the Body to the Brain
Although amyloidosis has faded from view, it has not entirely disappeared. There remain a few active centers of excellence for the study of amyloidosis, such as the one established at Boston University Medical Center, and sporadic cases of the disease affecting the heart, lung or peripheral nerves occasionally meet up with rheumatology consult services. For the most part though, the intense interest in the biology of amyloid has shifted from the body to the brain, where it continues to gain interest as a major target of study among neuroscientists.
For good reason, amyloidosis is often linked with the work of the noted German neuropathologist and clinician, Alois Alzheimer, MD, whose paper, “On a Peculiar Severe Disease Process of the Cerebral Cortex,” presented at a conference in 1906, described a curious finding: the presence of neurofibrillary tangles in the cerebral cortex of a patient, Auguste D.
Cognitively normal people can accumulate a large amyloid load in their brains, so the simple accumulation of amyloid in the brain is insufficient to cause dementia. However, there appears to be a much clearer correlation between the number of neurofibrillary tangles containing tau found post-mortem & the degree of dementia observed in life.
Auguste D. was described as a 51-year-old delusional, forgetful, disoriented, anxious, suspicious, unruly and disruptive woman who exhibited a striking personality change over the course of one year. She died of pneumonia, and Dr. Alzheimer made these prescient post-mortem observations of her brain cells: “In the center of an otherwise almost normal cell there stands out one or several fibrils due to their characteristic thickness and peculiar impregnability.” Regarding the senile plaques that he also observed, he wrote: “Numerous small miliary foci are found in the superior layers. They are determined by the storage of a peculiar material in the cortex.”3
Yet the discovery of amyloid predates this astute characterization. In 1853, a decade before Dr. Alzheimer’s birth, another German professor and physician, Rudolf Virchow, MD, identified a form of amyloid when he examined deposits of this seemingly abnormal extracellular material in liver tissue and described how it reacted with iodine and sulfuric acid by turning blue, confirming the presence of starch. He coined the term “amyloid,” based on the Greek word amylon, meaning starch.4
So what, after all, is amyloid? The amyloidoses comprise a group of protein-misfolding diseases that can complicate chronic inflammatory disorders, such as rheumatoid arthritis, or chronic infections (AA amyloidosis) or may develop in the setting of excess light chain production in patients with multiple myeloma (AL amyloidosis).
Amyloid fibrils are derived from the acute-phase reactant serum amyloid A (SAA) protein, an apolipoprotein constituent of high-density lipoprotein that is synthesized by hepatocytes under the transcriptional regulation of pro-inflammatory cytokines, particularly interleukin-6.2 The fibrils are composed of proteins that often have been mutated, partially fragmented or otherwise altered, which predisposes them to adopt an abnormal conformation.
Sustained overproduction of SAA is a prerequisite for the development of AA amyloidosis; although for reasons that are poorly understood, amyloidosis occurs only in a small proportion of patients with chronic inflammatory disorders. Interestingly, in Simone’s case, we never identified an underlying inflammatory disease.
The Amyloid Hypothesis
In the brain, amyloid precursor protein is cleaved to form amyloid beta (Aβ), a 40-amino-acid-long peptide that can aggregate in neurons to form soluble structures.5 However, when improperly folded, the tangled protein gets tossed into the cell, creating a bit of a mess, which in cell biology is never a good thing, because these structures steadily accumulate inside the cell. Lacking mechanisms for their proper disposal, the tangles become toxic to nerve cells. Over time, as more and more neurons become afflicted and then impaired, memory loss and personality changes ensue, leading to the clinical and pathological state so elegantly described by Dr. Alzheimer more than a century ago.
This concept of amyloid accumulation forms the basis for what is known as the amyloid hypothesis, the predominant framework for the pathogenesis of Alzheimer’s disease (AD). Aβ plaques are thought to wreak havoc by blocking proteasome function, inhibiting mitochondrial activity, altering intracellular Ca2+ levels and stimulating inflammatory processes.
Most importantly, Aβ plaques interact with the signaling pathways that regulate the phosphorylation of the microtubule-associated protein tau. Hyperphosphorylation of tau disrupts its normal function in regulating axonal transport and leads to the accumulation of neurofibrillary tangles and toxic species of soluble tau.5 In fact, accumulation of tau, rather than amyloid, may be the true culprit for the development of cognitive impairment.
The intense interest in the biology of amyloid has shifted from the body to the brain, where it continues to gain interest as a major target of study among neuroscientists.
Cognitively normal people can accumulate a large amyloid load in their brains, so the simple accumulation of amyloid in the brain is insufficient to cause dementia. However, there appears to be a much clearer correlation between the number of neurofibrillary tangles containing tau found post-mortem and the degree of dementia observed in life.6 Thus, a new view proposes that Aβ plaque formation is the key initiator of a complex pathogenic cascade that causes AD.
Aβ plaques act primarily as a trigger of other downstream processes, particularly tau aggregation, which mediate neurodegeneration. The major pathogenic effects of Aβ plaques may occur very early in the disease process.7 There may be little benefit in attacking these plaques later in their development. This suspicion has been confirmed by the disappointing results of several clinical trials.8 Targeting critical enzyme inhibitors of plaque formation has failed, too, and a trial using aggregated human Aβ as a therapeutic agent was halted when some participants developed an autoimmune encephalopathy.9 It may be time to abandon the amyloid hypothesis and seek a better explanation for the neuronal destruction caused by AD.
An Unrelenting Adversary or An Unanticipated Ally?
Unfortunately, the deposition of amyloid in any tissue, especially the brain, never relents. The high prevalence of AD has affected countless families as it progressively destroys the brains of some of our loved ones. But it is a condition that is not limited to the aged.
More recently, the term chronic traumatic encephalopathy (CTE) has been applied when the neuropathology findings of boxer’s brain were observed in retired professional football and hockey players, entertainment wrestlers, victims of domestic violence and military veterans exposed to blast and concussive injuries from improvised explosive devices. The cognitive and behavioral symptoms of CTE begin insidiously; first, there may be mood changes such as depression, apathy and irritability, which sadly may end in suicide.11 CTE is characterized by prominent deposition of tau and variable degrees of diffuse amyloid deposition, although the areas of brain involvement seem to differ from what is seen in AD.
Could there be any benefit to amyloid’s presence in our bodies? There may be a silver lining. Unlike other human biological fluids, healthy semen contains multiple types of amyloid fibrils. In a recent study, it was noted that these fibrils inhibited fertilization by immobilizing sperm.12 Interestingly, this immobilization facilitated the uptake and clearance of sperm by macrophages, which are known to infiltrate the female reproductive tract following semen exposure. In the presence of semen fibrils, damaged and apoptotic sperm were more rapidly phagocytosed than healthy ones, suggesting that the deposition of semen fibrils in the female reproductive tract facilitates the clearance of poor-quality sperm.
It’s heartening to learn that nature has found a way to put this pernicious protein to proper use somewhere in our bodies. Who knew? Amyloid as sperm selector; amyloid as our ally.
 Simon M. Helfgott, MD, is associate professor of medicine in the Division of Rheumatology, Immunology and Allergy at Harvard Medical School in Boston.
Simon M. Helfgott, MD, is associate professor of medicine in the Division of Rheumatology, Immunology and Allergy at Harvard Medical School in Boston.
References
- Patel VG, Kalakuntla V, Fortson JK, et al. Stercoral perforation of the sigmoid colon: Report of a rare case and its possible association with nonsteroidal anti-inflammatory drugs. Am Surg. 2002 Jan;68(1):62–64.
- Lachmann HJ, Goodman HJB, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007 Jun 7;356(23):2361–2371.
- Ramirez-Bermudez, J. Alzheimer’s disease: Critical notes on the history of a medical concept. Arch Med Res. 2012 Nov;43:595–599.
- George DR, Whitehouse PJ, D’Alton S, et al. Through the amyloid gateway. Lancet. 2012 Dec 8;380(9858):1986–1987.
- Dawkins E, Small DH. Insights into the physiological function of ß amyloid precursor protein: Beyond Alzheimer’s disease. J Neurochem. 2014 Jun;129(5):756–769.
- Goedert M, Spillantini MG, Crowther RA. A Brief history of tau. Clin Chem. 2015 Nov;61(11):1417–1418.
- Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen.’ Nat Neurosci. 2015 Jun;18(6):800–806.
- Drachman DA. The amyloid hypothesis, time to move on: Amyloid is the downstream result, not cause, of Alzheimer’s disease. Alzheimers Dement. 2014 May;10(3):372–380.
- Gilman S, Koller M, Black RS, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005 May 10;64(9):1553–1562.
- Omalu BI, DeKosky ST, Minster RL, et al. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. 2005 Jul;57(1):128–134; discussion 128–134.
- McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013 Jan;136(Pt 1):43–64.
- Roan NR, Sandi-Monroy N, Kohgadai N, et al. Semen amyloids participate in spermatozoa selection and clearance. Elife. 2017 Jun 27;6. pii: e24888.