 From Scripture’s women to New World missionaries (and the doctors in between), the first description of psoriasis as a clinical entity is disputed, but it can surely be attributed to either the Old Testament or Torah (Tzaarat, possibly afflicting Miriam), or the Roman encyclopedist Cornelius Celsus (25 BCE to 50 AD).1 What’s certain is that at least 15 centuries separated Galen coining the term psoriasis from the first recorded characterization of concomitant cutaneous and joint involvement afflicting Fray Pedro de Urraca, a Spanish Mercedarian monk living in Peru in the late 1600s.
From Scripture’s women to New World missionaries (and the doctors in between), the first description of psoriasis as a clinical entity is disputed, but it can surely be attributed to either the Old Testament or Torah (Tzaarat, possibly afflicting Miriam), or the Roman encyclopedist Cornelius Celsus (25 BCE to 50 AD).1 What’s certain is that at least 15 centuries separated Galen coining the term psoriasis from the first recorded characterization of concomitant cutaneous and joint involvement afflicting Fray Pedro de Urraca, a Spanish Mercedarian monk living in Peru in the late 1600s.
It would take another two centuries and a couple of brilliant French dermatologists to first make this association in Europe and later conceive the phrase psoriasis arthritique.2,3
This historical gap in medical taxonomy is akin to the temporal lag that characterizes a psoriatic arthritis (PsA) patient’s journey, typically affecting the skin for several years until clinically apparent synovio-entheseal involvement ensues. In fact, psoriasis is arguably the only known preclinical inflammatory arthritis condition that has an established, self-evident, clinical risk factor (i.e., biomarker) for disease development. As such, this represents a unique opportunity to study the dynamic events occurring during subclinical arthritis in general and provides the potential for early intervention in PsA (as well as prevention) in a susceptible and readily identifiable population.
The ability to recognize those who are predestined to develop PsA, however, is hindered by an incomplete understanding of the phenotypic, environmental, molecular and cellular events governing the transition from cutaneous to joint inflammation.
It is by unraveling the fundamental enigma of why (and when) one in four people with psoriasis goes on to develop extra-cutaneous joint inflammation that the field will most likely make the next leap in scientific discovery.
The Role of Genetics
Although genetic susceptibility plays a significant role in disease pathogenesis, it only partially accounts for the development of psoriatic-disease clinical manifestations. This includes the high familial aggregation for psoriatic disease, the discovery that certain human leukocyte antigen (HLA) alleles correlate with specific PsA phenotypes and the identification of numerous non-MHC (major histocompatibility complex) risk factors.4,5
Importantly, however, studies in monozygotic twins reveal a substantial proportion of discordance in both psoriasis (33%) and PsA (only 11%!), highlighting the influence that epigenetics and environmental factors can impose on disease development and perpetuation.6,7,8
Potential Triggers
Trauma & microbes offer some clues to PsA pathogenesis
The fact that non-genetic factors play a prominent role in the clinical manifestations of PsA is not surprising because previous research has revealed immune system variation to be determined by mostly non-heritable influences.9 The quest for understanding these environmental triggers and immunologic determinants of PsA development and perpetuation has been pursued.
One of the better studied instigators was first characterized by Heinrich Koebner (1876), who reported that mechanical trauma induced the development of new psoriatic lesions in nonlesional skin of patients with psoriasis.10 A deep Koebnerization phenomenon has also been extensively described for PsA.11 This mechano-inflammation hypothesis is supported by both the temporal association between injury and onset of the exaggerated inflammatory response, as well as the favored anatomical localization to structures exposed to micro- or macrotrauma, including the entheses and the joints. It can also justify the asymmetric, oligoarticular distribution of synovio-entheseal inflammation in PsA.12
One of the most intriguing potential triggers for psoriatic disease is the microbiome, the collection of symbiotic, commensal and pathogenic microorganisms (and their genomes) that reside in mammalian skin, mucosal surfaces and other organs. The adult human microbiome is for the most part robust and resilient. However, the perturbation of this equilibrium (i.e., dysbiosis) has been implicated in the initiation and perpetuation of several immune-mediated disorders, including psoriatic disease.13,14
In fact, studies in both animal models and humans have demonstrated a mechanistic link between the microbiome, gut inflammation and psoriatic disease. For example, multiple established murine models using mice that are prone to the development of psoriasiform rash and spondyloarthritis features do not demonstrate the phenotype under germ-free conditions but exhibit an interleukin 23 (IL-23)/IL-17-driven, spondyloarthritis-like inflammatory process when exposed to specific microbes, hence strengthening the hypothesis that intestinal dysbiosis is required to orchestrate synovioentheseal and cutaneous disease.15-17
In fact, multiple studies in humans have also demonstrated perturbations in the intestinal microbiota composition in psoriatic disease, as well as worsening pathology after fecal microbial transplantation.14,18-21 The prevailing hypothesis posits that this dysbiotic state leads to dysregulation of the gut epithelial barrier and the stimulation of IL-23-producing resident immune cells.22 IL-23-responsive mucosal innate cells are then activated with the capacity to circulate away from the gut and migrate to the enthesis and synovial tissues to cause inflammatory responses.23,24
Immune Mediators
From insult to clinical phenotype
The most remarkable evidence that unique cells are both necessary and sufficient in pathogenesis derive from the anecdotal—but compelling—observations that psoriatic disease can be (and has been) effectively cured in humans. In fact, several cases illustrated in the literature describe how unilateral, central and peripheral neurogenic disorders, such as stroke and post-poliomyelitis paralysis, spare the affected limb from psoriatic skin and joint inflammation, suggesting the need for neuronal signals to trigger local immune responses.25-27
The underlying mechanisms that can explain this brain-psoriatic axis are slowly emerging. For example, in an animal model of psoriasis it was found that psoriasiform rash was dependent on the interaction between a subset of sensory neurons and skin-resident dermal dendritic cells (DCs).28 Intriguingly, upon selective ablation of these nociceptors, these dermal DCs fail to produce IL-23 and the subsequent recruitment to the skin of IL-17-producing γδ T cells.
These findings suggest a scenario in which pain fibers integrate environmental signals to modulate local immune responses to a variety of microbial and pro-inflammatory stimuli in psoriatic disease, possibly explaining the Koebnerization phenomenon.29 This was also demonstrated in murine arthritis models, where knee denervation protected against joint inflammation, as well as in humans, where studies stimulating the vagus-nerve system showed inhibition of cytokine production and attenuation of disease severity in rheumatoid arthritis (RA).30,31
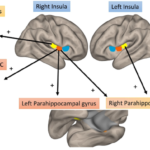
Functional brain magnetic resonance imaging has emerged as a novel tool to measure brain connectivity in inflammation. Its recent use in RA demonstrated that increased connectivity between areas implicated in self-referential mental activity (i.e., default mode network) and multimodal sensory processing (i.e., the insula cortex) was associated with increased RA disease activity and rates of centralized pain.32 Moreover, inflammation correlated with increased connectivity in brain regions involved in sensory input, memory and reward pathways.33
Whether or not these approaches will bring further insights and/or demonstrate therapeutic potential in PsA remains to be seen, but they provide proof-of-principle that the neuroimmune connection is of high relevance for modulating synovitis in psoriatic and related diseases. Notably, it has been demonstrated that the transferring of a whole new population of bone marrow-derived cells from a healthy donor to an affected histoidentical host or receiving an allogeneic hematopoietic stem cell transplantation can resolve both psoriasis and PsA, leading to drug-free remission for up to 20 years in some cases.34,35
Consequently, if replacing the entire set of immune cells can all but cure psoriatic disease, the question in the field has been one of specificity: Which particular cells (or group of cells) are responsible for disease pathogenesis?
Over the years, it became clear that innate and adaptive immune cells were responsible, at least partially, for the immune-mediated psoriatic syndrome. Among the most significant advances in our appreciation of the pathogenesis of PsA is the central role played by the pro-inflammatory subset of CD4+ T cells known as Th17 cells.36 These cells are activated by IL-23 to secrete the cytokines IL-17A, IL-17F and IL-22, which act on resident surrounding epithelial and endothelial cells to elicit the production of inflammatory cytokines and chemokines, often leading to the recruitment of other inflammatory cells and activation of innate epithelial defense mechanisms.37
In particular, elevation of Th17 cell subsets has been observed in peripheral blood, skin and joints of patients with psoriatic disease, and biologic therapies targeting both IL-23 and IL-17 are now U.S. Food and Drug Administration-approved for the treatment of moderate to severe psoriasis and joint disease.38-41
As discussed, other Th17 cells participate in psoriatic inflammation and have been shown to differentiate PsA from other arthritides (e.g., rheumatoid arthritis). Specifically, γδ-IL-23R+, IL-17-producing entheseal resident cells have been found to drive inflammation in an animal model of PsA-like disease.42 As in the gut, it was also reported that CD8-T cells and ILC-3 cells, both of which also release IL-17, are expanded in psoriatic but not rheumatoid joint fluid.43
In humans, the role of CD8+ T cells has been long recognized.44 In fact, the number of these cells significantly correlates with both disease activity and inflammatory findings on musculoskeletal ultrasound. With the advent of single cell sequencing technologies, clonal expansions of CD8+ T cells in PsA synovial fluid were identified, some of which were also deemed tissue-resident memory T cells and characterized by their capacity to produce multiple proinflammatory cytokines (i.e., IL-17, tumor necrosis factor [TNF] and interferon-γ).45,46
Similarly, transcriptomic analyses comparing skin and joint biopsies demonstrated a predominant Th17 profile in psoriatic skin, and psoriatic synovial tissues exhibited strong signals in TNF and angiogenic pathways.47,48 Altered bone remodeling in PsA is characterized by dysfunctional osteoblast and osteoclast activity, which can lead to concomitant erosions and osteoproliferation within the same person. Multiple studies revealed that both IL-17 and TNF are important in driving this abnormal bone turnover, which is at least partially driven by an increase in the osteoclast precursor population, that could serve as yet another distinctive target for arthritis treatment and prevention.49,50
Despite this accumulating body of knowledge, critical gaps in our understanding of PsA etiology and the triggers behind TNF-producing cell activation, Type-17 cell expansion and the yet to be uncovered mediators of skin and joint inflammation greatly hinder our ability to identify pre-clinical arthritis in psoriasis patients.
Disruptive Thinking & the Future of PsA
Although the past 20 years have brought transformational advances in pathogenesis and therapeutics for both psoriasis and PsA, joint outcomes have consistently trailed those witnessed in the skin.51 The arrival of anti-TNF agents, followed by antibodies that target the IL-23/IL-17 axis, have radically improved our ability to treat psoriasis. Remarkably, a significant number of patients can now achieve total clearance of skin disease.52
However, this extraordinary degree of responses observed in the skin domain has not been attained in PsA, where up to half the patients do not experience clinically consequential synovio-entheseal improvement with blockade of TNF or IL-23/IL-17 pathways.41,53,54 Hence, and although the process is known to be reversible (i.e., via denervation or bone marrow transplantation), highly effective treatment strategies for PsA continue to remain unattainable for many.
Innovation (and perhaps even disruptive medical thinking) is of the essence—unraveling yet unidentified therapeutic targets, implementing combination therapy strategies with currently available modalities, as well as intervention tactics to halt PsA before it even becomes clinically evident.55,56
Fortunately, many of these initiatives have already launched and are actively being pursued. Prominent examples include such endeavors as the National Institutes of Health-Accelerating Medicines Partnership (AMP) Autoimmune and Immune Mediated (AIM) Diseases and the European Innovative Medicines Initiative (IMI), which integrate public-private partnerships to uncover new targets for diagnosis and therapeutics in psoriatic disease.57
These, combined with programs that tackle multi-therapy interventional approaches (for this multi-domain disease) and outright preventive strategies, promise to propel the field forward by studying the psoriasis to PsA continuum in an integrated and ever more detailed (both molecularly and phenotypically) manner.58,59
The hope, of course, is to better understand (and perhaps be able to interrupt) the biological features linking Miriam’s snowflakes and Fray Urraca’s hooked hands.
 Rebecca H. Haberman, MD, a recipient of the 2021 National Psoriasis Foundation Outstanding Young Investigator Award, is an instructor of medicine, Division of Rheumatology, New York University (NYU) Grossman School of Medicine and associate director of the NYU Langone Psoriatic Arthritis Center (NYU PAC).
Rebecca H. Haberman, MD, a recipient of the 2021 National Psoriasis Foundation Outstanding Young Investigator Award, is an instructor of medicine, Division of Rheumatology, New York University (NYU) Grossman School of Medicine and associate director of the NYU Langone Psoriatic Arthritis Center (NYU PAC).
 Jose U. Scher, MD, a recipient of the ACR Henry Kunkel Early Career Investigator Award, is the Steere-Abramson associate professor of medicine and the associate director of research and translational medicine, Division of Rheumatology, New York University (NYU) Grossman School of Medicine, and director of the NYU Langone Psoriatic Arthritis Center (NYU PAC) and Microbiome Center for Rheumatology and Autoimmunity (MiCRA).
Jose U. Scher, MD, a recipient of the ACR Henry Kunkel Early Career Investigator Award, is the Steere-Abramson associate professor of medicine and the associate director of research and translational medicine, Division of Rheumatology, New York University (NYU) Grossman School of Medicine, and director of the NYU Langone Psoriatic Arthritis Center (NYU PAC) and Microbiome Center for Rheumatology and Autoimmunity (MiCRA).
Disclosures
Jose U. Scher, MD, received consulting fees from: Janssen, Novartis, Pfizer, Sanofi, UCB, Amgen and AbbVie; research funding from the National Institutes of Health, the National Psoriasis Foundation, Beatrice Snyder Foundation, Bloomberg Philanthropies, Janssen and Pfizer.
Rebecca H. Haberman, MD, received consulting fees from Janssen; research funding from the National Institutes of Health, the National Psoriasis Foundation, the Rheumatology Research Foundation, Beatrice Snyder Foundation, Bloomberg Philanthropies, Janssen and Pfizer.
References
- Glickman FS. Lepra, psora, psoriasis. J Am Acad Dermatol. 1986 May;14(5 Pt 1):863–866.
- Alibert J-L. Précis théorique et pratique sur les maladies de la peau … par M. Alibert. Paris: Caille et Ravier. 1822.
- Bazin E, Sergent L. Leçons théoriques et cliniques sur les affections cutanées de nature arthritique et dartreuse considérées en ellesmême et dans leurs rapports avec les éruptions scrofuleuses, parasitaires et syphilitiques réd. et publ. par Lucien Sergent. Paris: A. Delahaye. 1860.
- Li Q, Chandran V, Tsoi L, et al. Quantifying differences in heritability among psoriatic arthritis (PsA), cutaneous psoriasis (PsC) and psoriasis vulgaris (PsV). Sci Rep. 2020 Mar 18;10(1):4925.
- Winchester R, Minevich G, Steshenko V, et al. HLA associations reveal genetic heterogeneity in psoriatic arthritis and in the psoriasis phenotype. Arthritis Rheum. 2012 Apr;64(4):1134–1344.
- Lønnberg AS, Skov L, Skytthe A, et al. Heritability of psoriasis in a large twin sample. Br J Dermatol. 2013 Aug;169(2):412–416.
- Grjibovski AM, Olsen AO, Magnus P, et al. Psoriasis in Norwegian twins: Contribution of genetic and environmental effects. J Eur Acad Dermatol Venereol. 2007 Nov;21(10):1337–1343.
- Gervin K, Vigeland MD, Mattingsdal M, et al. DNA methylation and gene expression changes in monozygotic twins discordant for psoriasis: Identification of epigenetically dysregulated genes. PLoS Genet. 2012 Jan;8(1):e1002454.
- Brodin P, Jojic V, Gao T, et al. Variation in the human immune system is largely driven by non-heritable influences. Cell. 2015 Jan;160(1–2):37–47.
- Kobner H. Zur atiologie der psoriasis. Vjschr Dermatol. 1876;3:559.
- Thorarensen SM, Lu N, Ogdie A, et al. Physical trauma recorded in primary care is associated with the onset of psoriatic arthritis among patients with psoriasis. Ann Rheum Dis. 2017 Mar;76(3):521–525.
- Ritchlin CT, Colbert RA, Gladman DD. Psoriatic Arthritis. N Engl J Med. 2017 May 25;376(21):2095–2096.
- Clemente JC, Manasson J, Scher JU. The role of the gut microbiome in systemic inflammatory disease. BMJ. 2018 Jan 8;360:j5145.
- Scher JU, Ubeda C, Artacho A, et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol. 2015 Jan;67(1):128–139.
- Manasson J, Blank RB, Scher JU. The microbiome in rheumatology: Where are we and where should we go? Ann Rheum Dis. 2020 Jun;79(6):727–733.
- Taurog JD, Richardson JA, Croft JT, et al. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med. 1994 Dec;180(6):2359–2364.
- Rehaume LM, Matigian N, Ormerod K, et al. IL-23 promotes fecal microbiota dysbiosis associated with susceptibility to spondyloarthritis and ileitis in ZAP-70 mutant SKG mice [abstract 1576]. Arthritis Rheumatol. 2017;69(suppl 10).
- Manasson J, Wallach DS, Guggino G, et al. Interleukin-17 inhibition in spondyloarthritis is associated with subclinical gut microbiome perturbations and a distinctive interleukin-25-driven intestinal inflammation. Arthritis Rheumatol. 2020 Apr;72(4):645–657.
- Alekseyenko AV, Perez-Perez GI, De Souza A, et al. Community differentiation of the cutaneous microbiota in psoriasis. Microbiome. 2013 Dec 23;1(1):31.
- Shapiro J, Cohen NA, Shalev V, et al. Psoriatic patients have a distinct structural and functional fecal microbiota compared with controls. J Dermatol. 2019 Jul;46(7):595–603.
- Kragsnaes MS, Kjeldsen J, Horn HC, et al. Safety and efficacy of faecal microbiota transplantation for active peripheral psoriatic arthritis: An exploratory randomised placebo-controlled trial. Ann Rheum Dis. 2021 Sep;80(9):1158–1167.
- Ciccia F, Bombardieri M, Principato A, et al. Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheum. 2009 Apr;60(4):955–965.
- Kenna TJ, Davidson SI, Duan R, et al. Enrichment of circulating interleukin-17-secreting interleukin-23 receptor-positive gamma/delta T cells in patients with active ankylosing spondylitis. Arthritis Rheum. 2012 May;64(5):1420–1429.
- Gracey E, Qaiyum Z, Almaghlouth I, et al. IL-7 primes IL-17 in mucosalassociated invariant T (MAIT) cells, which contribute to the Th17-axis in ankylosing spondylitis. Ann Rheum Dis. 2016 Dec;75(12):2124–2132.
- Veale D, Farrell M, Fitzgerald O. Mechanism of joint sparing in a patient with unilateral psoriatic arthritis and a longstanding hemiplegia. Br J Rheumatol. 1993 May;32(5):413–416.
- Wang TS, Tsai TF. Psoriasis sparing the lower limb with postpoliomyelitis residual paralysis. Br J Dermatol. 2014 Aug;171(2):429–431.\
- Weiner SR, Bassett LW, Reichman RP. Protective effect of poliomyelitis on psoriatic arthritis. Arthritis Rheum. 1985 Jun;28(6):703–706.
- Riol-Blanco L, Ordovas-Montanes J, Perro M, et al. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature. 2014 June 5;510(7503):157–161.
- Bordon Y. Neuroimmunology: A sensational IL-23 response. Nat Rev Immunol. 2014 Jun;14(6):354.
- Kane D, Lockhart JC, Balint PV, et al. Protective effect of sensory denervation in inflammatory arthritis (evidence of regulatory neuroimmune pathways in the arthritic joint). Ann Rheum Dis. 2005 Feb;64(2):325–327.
- Marsal S, Corominas H, de Agustín JJ, et al. Non-invasive vagus nerve stimulation for rheumatoid arthritis: A proof-of-concept study. Lancet Rheumatol. 2021 Apr;3(4):e262–e69.
- Basu N, Kaplan CM, Ichesco E, et al. Neurobiologic features of fibromyalgia are also present among rheumatoid arthritis patients. Arthritis Rheumatol. 2018 Jul;70(7):1000–1007.
- Schrepf A, Kaplan CM, Ichesco E, et al. A multi-modal MRI study of the central response to inflammation in rheumatoid arthritis. Nat Commun. 2018 Jun 8;9(1):2243.
- Kanamori H, Tanaka M, Kawaguchi H, et al. Resolution of psoriasis following allogeneic bone marrow transplantation for chronic myelogenous leukemia: Case report and review of the literature. Am J Hematol. 2002 Sep;71(1):41–44.
- Hinterberger W, Hinterberger-Fischer M, Marmont A. Clinically demonstrable anti-autoimmunity mediated by allogeneic immune cells favorably affects outcome after stem cell transplantation in human autoimmune diseases. Bone Marrow Transplant. 2002 Dec;30(11):753–759.
- Annunziato F, Cosmi L, Santarlasci V, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007 Aug 6;204(8):1849–1861.
- Korn T, Bettelli E, Oukka M, et al. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517.
- Leipe J, Grunke M, Dechant C, et al. Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum. 2010 Oct;62(10):2876–2885.
- Cauli A, Mathieu A. Th17 and interleukin 23 in the pathogenesis of psoriatic arthritis and spondyloarthritis. J Rheumatol Suppl. 2012 Jul;89:15–18.
- Leonardi C, Matheson R, Zachariae C, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Eng J Med. 2012 Mar 29;366(13):1190–1199.
- McInnes IB, Mease PJ, Kirkham B, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): A randomised, double-blind, placebocontrolled, phase 3 trial. Lancet. 2015 Sep 19;386(9999):1137–1146.
- Sherlock JP, Joyce-Shaikh B, Turner SP, et al. IL-23 induces spondyloarthropathy by acting on ROR-gammat+ CD3+CD4-CD8-entheseal resident T cells. Nat Med. 2012 Jul 1;18(7):1069–1076.
- Menon B, Gullick NJ, Walter GJ, et al. Interleukin-17+CD8+ T cells are enriched in the joints of patients with psoriatic arthritis and correlate with disease activity and joint damage progression. Arthritis Rheumatol. 2014 May;66(5):1272–1281.
- Costello P, Bresnihan B, O’Farrelly C, et al. Predominance of CD8+ T lymphocytes in psoriatic arthritis. J Rheumatol. 1999 May;26(5):1117–1124.
- Penkava F, Velasco-Herrera MDC, Young MD, et al. Single-cell sequencing reveals clonal expansions of pro-inflammatory synovial CD8 T cells expressing tissue-homing receptors in psoriatic arthritis. Nat Commun. 2020 Sep 21;11(1):4767.
- Steel KJA, Srenathan U, Ridley M, et al. Polyfunctional, proinflammatory, tissue-resident memory phenotype and function of synovial interleukin-17A+CD8+ T cells in psoriatic arthritis. Arthritis Rheumatol. 2020 Mar;72(3):435–447.
- Belasco J, Louie JS, Gulati N, et al. Comparative genomic profiling of synovium versus skin lesions in psoriatic arthritis. Arthritis Rheumatol. 2015 Apr;67(4):934–944.
- Nerviani A, Boutet M-A, Tan WSG, et al. IL-23 skin and joint profiling in psoriatic arthritis: Novel perspectives in understanding clinical responses to IL-23 inhibitors. Ann Rheum Dis. 2021 May;80(5):591–597.
- Ritchlin CT, Haas-Smith SA, Li P, et al. Mechanisms of TNF-alpha- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J Clin Invest. 2003 Mar;111(6):821–831.
- Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006 Nov 27;203(12):2673–2682.
- Scher JU. The 2018 landscape of RA, PsA, and SpA pathogenesis. Curr Opin Rheumatol. 2018 Jan;30(1):57–58.
- Armstrong AW, Siegel MP, Bagel J, et al. From the Medical Board of the National Psoriasis Foundation: Treatment targets for plaque psoriasis. J Am Acad Dermatol. 2017 Feb;76(2):290–298.
- Mease PJ, van der Heijde D, Ritchlin CT, et al. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis. 2017 Jan;76(1):79–87.
- Mease PJ, Gladman DD, Ritchlin CT, et al. Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: results of a double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2005 Oct;52(10):3279–3289.
- Ritchlin C, Scher JU. Strategies to improve outcomes in psoriatic arthritis. Curr Rheumatol Rep. 2019 Dec 7;21(12):72.
- Haberman RH, Castillo R, Scher JU. Induction of remission in biologic-naive, severe psoriasis and PsA with dual anticytokine combination. Rheumatology (Oxford). 2021 Jul 1;60(7):e225–e226.
- National Institutes of Health-Accelerating Medicines Partnership Autoimmune and Immune Mediated Diseases Program. https://www.niams.nih.gov/grants-funding/niams-supported-research-programs/accelerating-medicines-partnership-amp
- Scher JU, Ogdie A, Merola JF, et al. Moving the goalpost toward remission: The case for combination immunomodulatory therapies in psoriatic arthritis. Arthritis Rheumatol. 2021 Sep;73(9):1574–1578.
- Scher JU, Ogdie A, Merola JF, et al. Preventing psoriatic arthritis: Focusing on patients with psoriasis at increased risk of transition. Nat Rev Rheumatol. 2019 Mar;15(3):153–166.