Case Study
A 26-year-old female was referred to the rheumatology service two years after the insidious onset of “walking funny” that progressed to significant proximal muscle weakness over a 10-month period. Early in the course of her illness, her creatine kinase (CK) was 4,000. Subsequent muscle biopsy revealed muscle fiber degeneration and regeneration in addition to numerous phagocytic cells. She was diagnosed with polymyositis but, despite treatment with high-dose corticosteroids, methotrexate (MTX), and monthly intravenous immunoglobulin (IVIG), she did not improve. At the time of presentation to rheumatology, her CK was 8,000, and she reported continued progression of muscle weakness and consequent frequent falls. Her family history was remarkable for a sister with the onset of similar symptoms at 30 years of age. Because of the lack of response to previous appropriate therapies and the family history, we suspected a hereditary myopathy. Her initial muscle biopsy results were consistent with the possibility of a hereditary myopathy, because some dystrophies do show inflammation. Repeat muscle biopsy was recommended for more extensive histopathologic evaluation. Upon immunohistochemical staining, the absence of dysferlin was discovered, consistent with a diagnosis of limb-girdle muscular dystrophy 2B.

Introduction
This case highlights the difficulty in diagnosing polymyositis and the need to think through the appropriate differential diagnosis and to use the appropriate diagnostic modalities when evaluating patients with suspected idiopathic inflammatory myopathy (IIM). Polymyositis (PM), dermatomyositis (DM), and inclusion body myositis (IBM) comprise the major IIM subsets in adults. A further categorization based on associated malignancy or other connective tissue disease features is useful in some circumstances, such as directing therapeutic interventions. Although the IIMs share the characteristics of immune-mediated attacks on skeletal muscle resulting in muscle weakness, they are in fact heterogeneous diseases with varied histopathological and clinical characteristics.1 Furthermore, these inflammatory myopathies can be confused with other myopathies that sometimes can have an inflammatory component, such as muscular dystrophies.
Differential Diagnosis
The differential diagnosis of a patient with suspected IIM is vast (see Table 1, p. 19) and highlights the importance of a meticulous history and physical, as well as appropriate and thorough diagnostic testing. The still-useful classification criteria of Bohan and Peter require four of the following features for a diagnosis of definite PM or three features plus a characteristic rash for a diagnosis of definite DM:2,3
- Symmetric, subacute muscle weakness;
- Abnormal muscle biopsy: fiber size variation, necrosis, regeneration, atrophy, and inflammation;
- Muscle enzyme elevation: CK, aldolase, alanine transaminase (ALT), aspartate aminotransferase (AST), and lactic dehydrogenase (LDH);
- Electromyogram abnormality: triad of short, small, polyphasic motor units; insertional irritability, positive sharp waves, fibrillations; and bizarre, high-frequency repetitive discharges; and
- Typical DM skin abnormalities: Gottron’s papules (see Figure 2, below) or heliotrope rash.
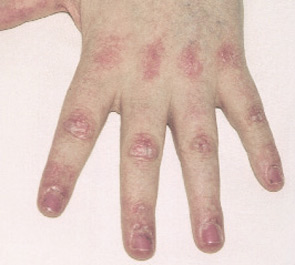
Exclusion criteria include evidence of metabolic, infectious, post-traumatic, and neuromuscular disorders.2,3 Although widely cited, Bohan and Peter’s criteria were developed prior to the discovery of myositis-specific autoantibodies (MSAs) and have been criticized for their inability to differentiate patients with PM, IBM, the toxic myopathies, and some genetic and metabolic myopathies.1
Incidence, Demographics, History, and Physical Exam
The collective incidence of the IIM is one in 100,000 per year. The female-to-male ratio for PM and DM is two to one; IBM occurs more frequently in males (three to one). Disease onset in PM is 18 or older; bimodal peaks of onset are seen in DM (5–15 and 45–65 years).4,5 IBM is most common in individuals greater than 50 years of age. However, it can occur earlier, particularly in familial cases. There are many masqueraders of IIM. See Table 2 (p. 19) for a list of characteristics that can point toward or away from a diagnosis of IIM.
Both PM and DM present subacutely over weeks to months with proximal muscle weakness. The photosensitive skin manifestations of DM include facial erythroderma without sparing of the nasolabial folds that is typical in lupus erythematosus; heliotrope rash around the eyes; Gottron’s rash/papules (see Figure 2, below); and shawl and v-neck signs. Patients with MSAs can be categorized by typical features associated with these antibodies. See Table 3 (p. 22) for a list of features associated with particular autoantibodies.
In contrast to the presentation of PM and DM, the onset and progression of IBM is characteristically slow, occurring over months to years. Asymmetric weakness, dysphagia, frequent falling, and distal as well as proximal muscle weakness and wasting are hallmarks of this IIM subset.
IIM can present in overlap with other rheumatic diseases, including systemic lupus erythematosus, progressive systemic sclerosis (scleroderma), Sjögren’s syndrome, and RA. The onset of myositis symptoms can occur at, before, or after diagnosis of these diseases. Overlap with IBM is far less common, but has been reported.

The association of IIM with malignancy is well recognized. In a review of four studies comprising 1,078 myositis patients and a comparable number of controls, the overall odds ratio (OR) for the association of cancer with DM was 4.4 (95%, confidence interval [CI] 3.0, 6.6) and with PM the OR was 2.1 (95% CI 1.4, 3.3).6 Malignancies are predominantly adenocarcinomas and usually develop within one year before or after myositis onset, but have been reported to occur up to five years later.7 Therefore, age-appropriate cancer screening and ongoing surveillance with a lowered threshold for further investigation is mandated.
Approximately 30% of DM cases present without clinically apparent muscle weakness, the majority of which progress months or years later to clinical myositis.8 Patients without evidence of muscle weakness or serum muscle enzyme abnormalities are classified as dermatomyositis sine myositis or amyopathic DM. These patients can develop interstitial lung disease (ILD) which can be rapidly progressive, heralding a poor prognosis.9,10
The importance of evaluation for toxic myopathies cannot be stressed enough, for as a group toxins represent the most common etiology of myopathy. In an elegant review of this topic, Walsh and Amato classified the toxic myopathies to date according to presumed pathogenic mechanism into seven categories:11
- Necrotizing myopathy: Alcohol, anti-lipemic agents (statins and fibrates), cyclosporin, labetolol, propofol, illicit drugs (cocaine, heroin, amphetamines), and controlled substances (meperidine, pentazocine);
- Amphiphilic: Amiodarone, chloroquine, and hydroxychloroquine;
- Antimicrotubular: Colchicine and vincristine;
- Mitochondrial myopathy: Zidovudine;
- Inflammatory myopathy: L-tryptophan, D-penicillamine, cimetidine, L-dopa, phenytoin, lamotrigine, interferon-α, hydroxyurea, and imatinib;
- Hypokalemic myopathy: Diuretics, laxatives, amphotericin, toluene abuse, licorice, corticosteroids, and alcohol abuse; and
- Unknown: Critical illness myopathy (corticosteroids, nondepolarizing neuromuscular blocking agents, and sepsis), omeprazole, isoretinoin, finasteride, and emetine.
Obtaining a family history of similar symptoms and neuromuscular disease is important; a positive response is most likely indicative of pathology other than IIM, such as in the muscular dystrophy in the case study above. A notable exception is IBM, because both idiopathic and familial forms exist.
Laboratory Tests
Muscle enzymes: As noted above, elevations of serum concentration of intracellular muscle components (CK, aldolase, AST, ALT, LDH, and myoglobin) generally accompany myositis. In IBM, serum CK correlates poorly with disease acuity and severity. It is normal in 25% of patients and, if abnormal, it is generally only slightly elevated.12
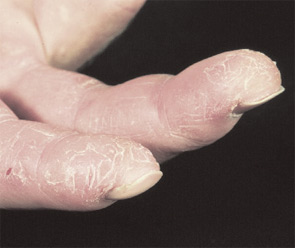
Tests commonly called “liver function tests” actually test for liver-associated enzymes such as AST, ALT, and LDH—which are also found in muscle. Unfortunately, it is common for patients to present to rheumatologists after a liver biopsy is performed due to elevated “liver function tests” resulting from active myositis. In this case, the liver biopsy is normal. Also remember that, when monitoring a patient with active myositis and elevated CK being treated with MTX, increased AST and ALT are more likely to reflect active disease than MTX-induced hepatic toxicity.
Autoantibodies: A positive antinuclear antibody titer is present in 60% to 80% of IIM patients; rheumatoid factor is positive in 8%.13 See Table 3 (p. 22) for a list of autoantibodies and associated features. These autoantibodies each are generally associated with a characteristic IIM phenotype. As is the case with all autoantibodies, positive and negative results must be interpreted in light of the clinical context, but a positive result can be useful prognostically as well as for guiding further evaluation and treatment.
EMG: As noted above, a myopathic EMG has a characteristic triad of features. New-onset disease can also result in an added feature of early recruitment. Although the EMG will not point to the specific etiologic entity (disease or toxin), it is helpful in ruling out neuropathic processes.
MRI: MRI has become an important diagnostic modality in IIM and serially may be useful in evaluating response to treatment in individual patients. Short tau inversion recovery images can demonstrate areas of edema which can be interpreted as active myositis (see Figure 1, p. 18). Although MRI should not substitute for muscle biopsy, it can guide muscle biopsy sampling by identifying sites of active disease. T1 MRI imaging is used to evaluate muscle atrophy and fatty replacement and thus serves as a measure of chronic muscle damage.
Muscle biopsy: Muscle biopsy is the essential and definitive diagnostic modality for IIM. Only very rarely should therapy proceed without a biopsy (one case is when a characteristic DM rash is present in a patient with a typical pattern of weakness but biopsy is unavailable). If immediate therapy is clinically deemed wise, a brief delay before biopsy will not significantly reduce the diagnostic usefulness because most biopsy signs recede slowly.
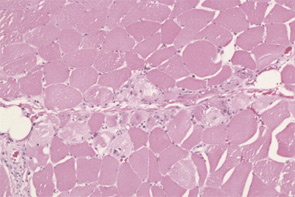
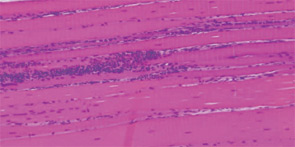
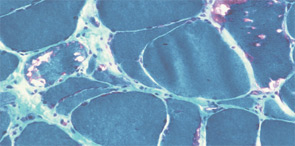
The pathogenesis of IIM appears to be reflected in the histochemical characteristics of each subset. DM is thought to result from humorally mediated vascular injury (see Figure 4A, p. 22). Perifascicular and perivascular inflammation and atrophy are most characteristic of DM, and vessel thrombosis, fiber necrosis, and regeneration can also be seen. In contrast, PM and IBM are thought to involve cytotoxic T cells in their pathogenesis. In PM, CD8-positive lymphocytes surround and invade muscle fibers expressing Major Histocompatibility (MHC) class I antigens (see Figure 4B, p. 22). IBM is likely to be present if the biopsy shows rimmed vacuoles containing basophilic granules and amyloid deposits in addition to inflammation (see Figure 4C, p. 22).
Treatment
An integral component of IIM treatment is rehabilitation medicine. Patients should be referred for evaluation and treatment by a physiatrist or physical therapist upon diagnosis. (See “Rehabilitation and Myositis,” p. 1, for more on physical therapy for myositis.) Active disease does not preclude patient-specific graded physical and occupational therapy that is combined with appropriate pharmacologic treatment.
Prednisone remains the first-line pharmacologic therapy for PM and DM. We advocate an initial dose of 1 mg/kg/day, with a pulse of methylprednisolone (typically 1 gm) at the outset in significantly affected patients. We favor disease modifying antirheumatic drug (DMARD) initiation (most often MTX at usual rheumatologic doses of up to 25 to 30 mg/week or azathioprine at up to 2 mg/kg/day) within a month of initiating prednisone in order to avoid disease flares while attempting to taper prednisone. If prednisone taper is not tolerated with single therapy of 25 to 30 mg MTX/week or 100 to 150 mg azathioprine/day, then consider a combination of azathioprine and MTX.14 In our experience using MTX as adjunct therapy for IIM, MTX-related pulmonary toxicity has been very uncommon.
Opinions vary considerably regarding the role of IVIG in IIM treatment. A controlled clinical trial has demonstrated efficacy in treatment-resistant DM and, in an open-label study, significant muscle strength improvement was reported in treatment-refractory PM.15,16 In our opinion, monthly IVIG infusions (1–2 gm/kg/month) should be considered for both DM and PM. Although many patients may benefit substantially, this effect often diminishes with a prolonged course of infusions. In cancer-associated myositis, when chemotherapy is used for the treatment of the tumor, IVIG can be a sensible option.
Added options in refractory DM and PM include cyclosporine and tacrolimus, particularly if there is co-existing interstitial lung disease.17,18 Early uncontrolled reports using mycophenylate mofetil have shown mixed results and no controlled trial has been reported. Because reports of cyclophosphamide efficacy in IIM are conflicting, this medication should be reserved for cases resistant to other immunosuppressants and IVIG.19
Despite some evidence for an immune contribution to the pathogenesis of IBM, and although some patients respond transiently to conventional IIM treatment, no therapy substantially affects the progression of this disease. However, no rigorous long-term trial has been reported.20 Significant improvement in dysphagia symptoms following IVIG has been reported.21
The role of anti–tumor necrosis factor or B cell–depleting therapies in IIM is unknown. Published uncontrolled case reports describe varied outcomes. Ongoing controlled clinical trials investigating the role of these agents in PM/DM may clarify the potential efficacy of these therapies.
Monitoring indices of active disease is important in guiding therapy and should not be limited to periodic measurement of serum muscle enzymes. The International Myositis Assessment and Clinical Studies (IMACS) international collaboration has developed a core set of three outcome measures for PM and DM: myositis disease activity, damage, and health-related quality of life.22,23 Although developed specifically for use in PM and DM clinical trials, these outcomes measures can be useful in clinical practice. The principle of comprehensive disease assessment is also applicable to IBM.

One of the most important principles of IIM treatment is that the question, “Is this IIM?”, should not be dismissed following initial diagnosis. Maintain a high index of suspicion and reassess the question in the following settings. When in doubt, re-biopsy.
- Persistent elevated muscle enzymes despite appropriate treatment and improving symptoms: Treat the patient, not the number! MRI can identify active inflammation in patients with conflicting indices of disease activity, such as persistent CK elevations without corresponding muscle weakness or vice versa;
- Persistent or increasing muscle weakness with or without CK change: This could represent active IIM, but a high index of suspicion for steroid myopathy or a high degree of muscle damage must be maintained; and
- Evolving symptoms suggestive of IBM in a patient whose muscle biopsy was read as PM: This probably is IBM, particularly in concert with appropriate clinical characteristics.
Conclusion
The IIMs are a heterogenous group of immune-mediated myopathies. Thorough consideration of the potential differential diagnosis and careful examination of a muscle biopsy are essential for accurate diagnosis and choice of appropriate therapy. Cornerstones of therapy are corticosteroids, DMARDS, and physiatry. If your myositis patient is not responding, rethink the diagnosis and consider appropriate consultations, laboratory studies, imaging, or re-biopsy.
Dr. Coyle is a clinical fellow in rheumatology at the National Institute of Arthritis, Skin and Musculoskeletal Diseases (NIAMS), National Institutes of Health (NIH) in Bethesda, Md. Dr. Plotz is chief of the arthritis and rheumatism branch of NIAMS/NIH. Dr. Gourley is program director of rheumatology fellowship training at NIAMS/NIH. This article was supported by the Intramural Research Program of NIAMS/NIH.
References
- Di Martino SJ, Kagen LJ. Newer therapeutic approaches: Inflammatory muscle disorders. Rheum Dis Clin North Am. 2006;32:121-128, ix.
- Bohan A, Peter J. Polymyositis and dermatomyositis (first of two parts). New Engl J Med. 1975;(292):344-347.
- Bohan A, Peter J. Polymyositis and dermatomyositis (second of two parts). New Engl J of Med. 1975;(292):403-407.
- Flachenecker P. Epidemiology of neuroimmunological diseases. J Neurol. 2006;253 [Suppl 5], V/2-V/8.
- Christopher-Stine L, Plotz PH. Adult inflammatory myopathies. Best Pract Res Clin Rheumatol. 2004;18(3):331-344.
- Zantos D, Zhang Y, Felson D. The overall and temporal association of cancer with polymyositis and dermatomyositis. J Rheumatol. 1994;21(10):1855-1859.
- Levine SM. Cancer and myositis: New insights into an old association. Curr Opin Rheumatol. 2006;18(6):620-624.
- Gerami P, Schope JM, McDonald L, Walling HW, Sontheimer RD. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis sine myositis): A missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006;54(4):597-613.
- Suda T, Fujisawa T, Enomoto N, et al. Interstitial lung diseases associated with amyopathic dermatomyositis. Eur Respir J. 2006;28(5):1005-1012.
- Ideura G, Hanaoka M, Koizumi T, et al. Interstitial lung disease associated with amyopathic dermatomyositis: Review of 18 cases. Respir Med. 2007;101(7):1406-1411.
- Walsh RJ, Amato AA. Toxic myopathies. Neurol Clin. 2005;23(2):397-428.
- Wortmann RL. Inflammatory Diseases of Muscle and Other Myopathies. In: Harris ED, Ruddy S, Budd RC, et al., eds. Kelly’s Textbook of Rheumatology, Seventh Edition. 2, 1309-1335. 2005. Philadelphia PA, Elsevier Saunders.
- Targoff IN. Laboratory testing in the diagnosis and management of idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 2002;28(4):859-890, viii.
- Villalba L, Hicks JE, Adams EM, et al. Treatment of refractory myositis: A randomized crossover study of two new cytotoxic regimens. Arthritis Rheum. 1998; 41(3):392-399.
- Dalakas MC, Illa A, Dambrosia JM, et al. A controlled trial of high dose intravenous immune globulin infusions as treatment for dermatomyositis. New Engl J Med. 1993;329:1993-2000.
- Cherin P, Herson S, Wechsler B, et al. Efficacy of intravenous gammaglobulin therapy in chronic refractory polymyositis and dermatomyositis: An open study with 20 adult patients. Am J Med. 1991;91(2):162-168.
- Oddis CV, Sciurba FC, Elmagd KA, Starzl TE. Tacrolimus in refractory polymyositis with interstitial lung disease. Lancet. 1999;22;353(9166):1762-1763.
- Ytterberg SR. Treatment of refractory polymyositis and dermatomyositis. Curr Rheumatol Rep. 2006;8(3):167-173.
- Cordeiro AC, Isenberg DA. Treatment of inflammatory myopathies. Postgrad Med J. 2006;82(969):417-424.
- Griggs RC. The current status of treatment for inclusion-body myositis. Neurology. 2006;24;66(2 Suppl 1):S30-S32.
- Cherin P, Pelletier S, Teixeira A, et al. Intravenous immunoglobulin for dysphagia of inclusion body myositis. Neurology. 2002;58(2):326.
- Miller FW, Rider LG, Chung YL, et al. Proposed preliminary core set measures for disease outcome assessment in adult and juvenile idiopathic inflammatory myopathies. Rheumatology (Oxford). 2001;40:1262-1273.
- IMACS Web site. https://dir-apps.niehs.nih.gov/imacs/index.cfm?action=home.main. Last accessed December 4, 2007.