Advances in CPPD Pathogenesis and Molecular Genetics
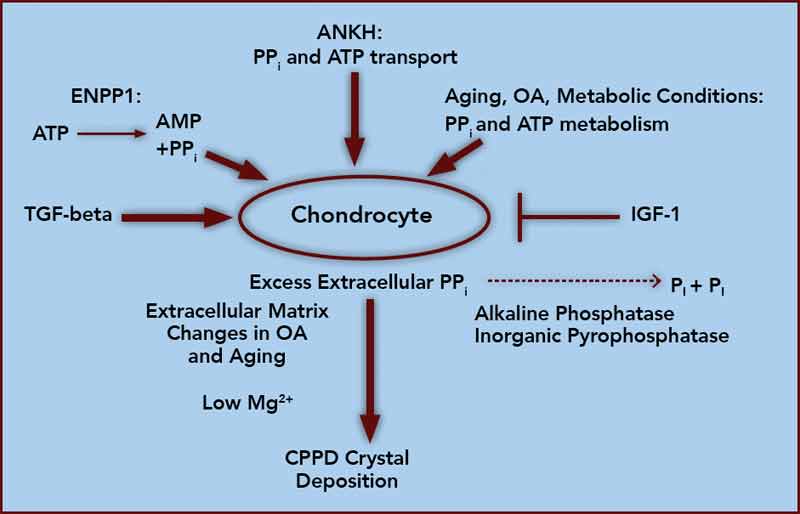
At the core of crystal deposition in idiopathic and other forms of CPPD is dysregulated PPi metabolism, accompanied by altered chondrocyte differentiation and function (see Figure 1).7,8 Articular cartilage, unlike growth-plate cartilage, is specialized to avoid the process of matrix calcification. Relatively copious output of PPi by chondrocytes physiologically functions to limit basic calcium phosphate crystal growth. This protective mechanism becomes deleterious by excessive PPi output in CPPD.7,8 Factors responsible for upregulating PPi generation in the aging joint and in OA are likely heterogeneous, but most function through increasing articular ectonucleotide pyrophosphatase phosphodiesterase (ENPP1) and the multiple-pass, transmembrane protein ankylosis protein homolog human gene (ANKH).7,8
ENPP1 generates intracellular and extracellular PPi via hydrolysis of ATP and other nucleoside triphosphates, and articular ecto–nucleotide pyrophosphatase/phosphodiesterase (ecto-NPP) activity is substantially elevated in both CPPD and OA.8 Intracellular PPi and ATP are transported to the extracellular space by processes involving ANKH, whose expression rises in OA chondrocytes.7,8