click for large version
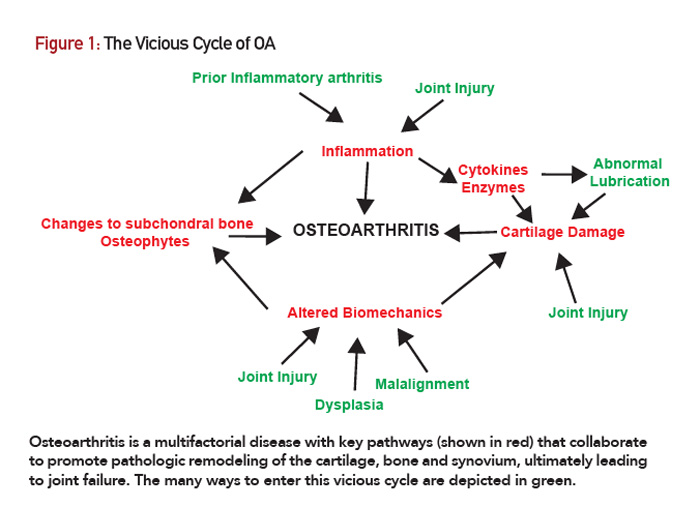
Figure 1: The Vicious Cycle of OA
Osteoarthritis is a multifactorial disease with key pathways (shown in red) that collaborate to promote pathologic remodeling of the cartilage, bone and synovium, ultimately leading to joint failure. The many ways to enter this vicious cycle are depicted in green.
Enzyme Inhibitors
The concerted effort of proteases is thought to mediate the slow degradation of articular cartilage extracellular matrix observed in OA.8 These proteases include zinc-dependent enzymes, such as MMP13 and the aggrecanases (e.g., ADAMTS5), as well as cysteine proteases, such as cathepsin K.