
The current therapies available for treatment to prevent fracture from osteoporosis target pathways of bone remodeling. We need to understand the mechanisms of bone resorption and bone formation, and the cells that control these mechanisms in order to have an impact on fracture prevention.
Osteoclast Development
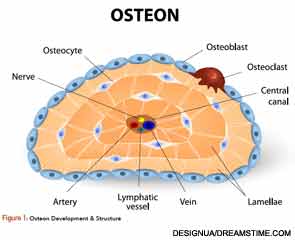
Osteoclasts (see Figure 1) are derived from hematopoietic stem cells, which when influenced by certain stimulatory factors, such as
macrophage-colony-stimulating factor (M-CSF), differentiate into monocytes. In the presence of receptor activator for nuclear factor kappa β ligand (RANKL) and M-CSF, they become multinucleated.
RANKL is essential for osteoclast formation. It is produced by osteoblasts and, ultimately, regulates bone remodeling.3 Osteoclasts attach to bone matrix, dissolving matrix and the organic part of bone, and producing tartrate-resistant acid phosphatase (TRAP), lysosomal enzymes, cathepsin K and integrins (see Figure 2). Osteoclasts express calcitonin receptors and RANK. This makes osteoclasts a major therapeutic target in osteoporosis.
To regulate osteoclast function, the formation, number, activity and lifespan of the osteoclast need to be controlled. This is accomplished through the RANK/RANKL/Osteoprotegerin (OPG) pathway. OPG is a cytokine receptor that is produced by osteoblasts and a member of the tumor necrosis factor (TNF) receptor family. OPG can decrease production of osteoclasts by inhibiting the differentiation of osteoclast precursors into osteoclasts and also regulate the resorption of osteoclasts in vitro and in vivo.
Osteoblasts, through RANKL and interleukin (IL) 1, IL-6 and TNF, activate osteoclasts to mature. When osteoclasts resorb, they release transforming growth factor beta (TGF-β) and insulin growth factors (IGF-1, IGF-2), which stimulate the osteoblast to mature and form bone.4
OPG production is stimulated in vivo by the female sex hormone estrogen, as well as the osteoporosis drug strontium ranelate.5 OPG has been used experimentally to decrease bone resorption in women with postmenopausal osteoporosis and in patients with lytic bone metastases. Another drug that can affect OPG is denosumab, because it can act as a decoy receptor for osteoblast-derived RANKL.
Denosumab increases bone mineral density (BMD) and reduces bone turnover markers (BTM) in postmenopausal women with low BMD and osteoporosis.6,7 These effects are reversible after discontinuation, and retreatment does produce as significant a response as before. Vertebral, hip and nonvertebral fractures are significantly reduced by treatment with denosumab.8 Studies demonstrate increases in BMD and decreases in BTMs with denosumab more so than with alendronate.9 Patients who switch therapy from alendronate to denosumab increase BMD more than those continuing on alendronate.10
When the osteoclast attaches to the bone surface, it releases acid, which demineralizes bone, releasing proteins that degrade nonmineralized tissue. Cathepsin K is a lysosomal cysteine protease with an important role in the function of osteoclasts. It acts to degrade bone collagen. By inhibiting cathepsin K, the removal of bone matrix is reduced. Clinical trials have demonstrated that cathepsin K has no effect on the function, formation or survival of the osteoclast.
Osteoblasts
Preosteoblasts, which are derived from mesenchymal stromal cells, differentiate into osteoblasts. These cells may get buried into bone and become osteocytes, or they can be lining cells on the surface of the bone or differentiate into mature osteoblasts. These bone formation cells stimulate matrix mineralization, make type I collagen, alkaline phosphatase, osteocalcin, fibronectin and collagenase. Receptors on the osteoblast surface, crucial for control of bone turnover and calcium homeostasis, include parathyroid hormone (PTH) receptors, vitamin D and TGF-β. RANKL is also expressed on the surface of the osteoblast and is essential to bone remodeling pathways, most specifically for osteoclastogenesis.
The Wnt signaling pathway is critical for regulation of cell growth, differentiation, function and death, and this is especially true in bone metabolism.11 The Wnt pathway has a frizzled and LDL receptor-related protein (LRP-5/6) co-receptor. The Wnt protein signals after attaching to its receptors, through beta catenin, genes that direct osteoblast maturation to turn on.
Wnt/LRP5/β-Catenin Pathway in Bone Formation
The Wnt/LRP5/β catenin pathway is essential for osteoblast proliferation, differentiation and survival.12 Loss of function mutations in LRP5 cause severe osteoporosis and fracture as seen in the osteoporosis pseudoglioma syndrome (OPPG), while gain of function mutation in LRP5 produces the high bone-mass “trait.”12 LRP5 in bone formation may be directed by signals from the gut (small intestine).13
Upregulation of serotonin acting vita receptors (subtype Htr1b) inhibits bone formation. Loss of signaling or production has a net bone remodeling effect and stimulates high bone mass.13
The current therapies available for treatment to prevent fracture from osteoporosis target pathways of bone remodeling.
Natural inhibitors of the Wnt/LRP5/β catenin pathway include dickkopf (DKK) made by myeloma cells; soluble decoy receptors, such as Wnt inhibitory factor (WIF); secreted frizzled-related protein (sFRP); and sclerostin (SOST) a product of osteocytes.
Sclerostin inhibits Wnt signaling, which causes inhibition of osteoblast function. Sclerostin antibody stimulates osteoblast function by allowing for increased Wnt signaling. Following a single dose of sclerostin antibody, there may be marked increases in markers of bone formation compared with markers of bone resorption.

Animal studies with knockout mice (SOST-/-) demonstrate greater than a 50% increase in BMD (lumbar spine and femur) with sclerostin antibody. Micro CT studies show increases in both trabecular and cortical bone. Histomorphometric analyses demonstrate greater than a nine-fold increase in the osteoblast surfaces, with no change in the osteoclast surface (uncoupling). Increased strength is demonstrated via mechanical testing. In ovariectomized rats, there was complete reversal of one year of estrogen-deficiency-induced bone loss. Bone mass and strength increased to levels that exceeded the results found in nonovariectomized controls.
Osteocytes
Osteocytes are derived from the osteoblast and are buried inside the bone. They are responsible for sensing mechanical loads in bone and sending signals to the surface and the interior of the bone via long projections from the cell body through the matrix and to the surface osteoblasts. They can synthesize matrix proteins. It has been documented that in the presence of steroids, there is increased apoptosis of the osteocyte.5 Ultimately, osteocytes may control remodeling through signaling to the bone surface and directing osteoclasts when and where to resorb, and osteoblasts where to form bone.
Metabolic Bone Effects of Biologics
Bone and cartilage loss in rheumatoid arthritis (RA) is measured by X-ray, ultrasound and magnetic resonance imaging. Disease modifying antirheumatic drugs and biologics can halt or retard the progression of joint destruction. IL-1 and tumor necrosis factor alpha (TNFα) have significant effects on bone remodeling.14
Focal bone loss in RA occurs through multiple mechanisms, including matrix metalloproteinase (MMP) via osteoclast precursor cells. Osteoclasts are the major cell types that mediate focal bone loss in RA.14 Patients with RA or inflammatory arthritis have more osteoclasts around the joint and in the bone than individuals with no arthritis, which causes resorption of bone and bony erosions, as well as bone loss in inflamed joints.15 Osteoclasts produce TRAP and cathepsin K, which work to degrade bone matrix. The expression of calcitonin receptors coincides with the differentiation of the osteoclast into a fully competent resorbing cell. Multinucleated cells expressing messenger RNA (mRNA) for TRAP, cathepsin K and the calcitonin receptor were found in resorption lacunae where pannus invaded bone. Other cell types, including activated synovial fibroblasts or macrophages, may contribute to focal bone erosions. RANKL is necessary for osteoclast differentiation and activation. Osteoprotegerin (OPG) acts as a decoy receptor to block RANKL and, ultimately, osteoclastogensis.
Sites at which regulatory factors control osteoclast differentiation and activity include bone-lining cells affected by PTH, PTH-related peptide (PTHrP—the humoral factor associated with hypercalcemia of malignancy), 1,25 OH2 Vit D3, prostaglandin E2, and cytokines IL-1, TNFα, IL-11 and IL-17. Factors including M-CSF, RANKL, IL-1, TNFα, IL-6, IL-11 and IL-15 act on osteoclast precursors to induce differentiation into osteoclasts.14
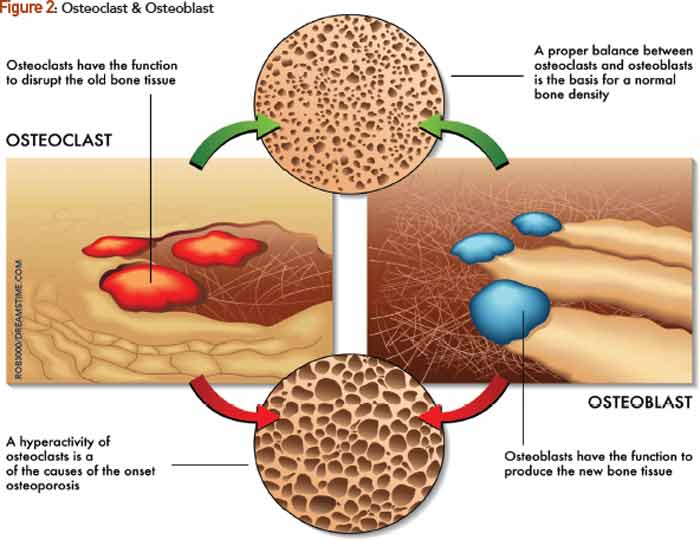
Several factors act on osteoclasts directly to enhance resorption, including IL-1, TNFα and RANKL. As demonstrated in multiple studies, RANKL is important in focal bone loss in RA. RANKL mRNA and protein are expressed in synovial tissue in patients. Expression is localized in synovial fibroblasts and T cells, which may induce osteoclastogenesis in vitro. Of note, however, is that treatment of cell cultures with OPG did not completely block osteoclastogenesis, as might be expected, which suggests that other proteins or factors from synovial cells may also have osteoclast-inducing potential.14
Cartilage destruction in RA can occur through direct release of products in pannus that degrade the matrix, specifically MMP and cathepsins. Indirect mechanism involves release of pannus-derived cytokines, which induce chondrocytes to release factors that degrade the matrix, including nitric oxide (NO), IL-1 and TNFα.14
There is compelling data to suggest that TNFα antagonists have an effect on NO production in human cartilage. Osteoarthritic cartilage (OA) produces NO, which is stimulated by proinflammatory cytokines TNFα, IL-1β, IL-17 and bacterial lipopolysaccharide.16
Both infliximab and etanercept, TNFα antagonists, inhibited TNFα-induced NO production in a dose-dependent mechanism with no evidence of any impact on IL-1β, IL-17 and LPS stimulated synthesis. Data from the study suggests that endogenous-cartilage-derived TNFα antagonists modulate NO production in the cartilage of OA patients. TNFα and soluble TNFα receptor (sTNFRI and sTNFRII), both made by human OA cartilage, when subjected to a neutralizing antibody, enhanced NO production against sTNFRI, but not against sTNFRII. This suggests that TNFα is not an autocoid (hormone-like substance) mediator in these functions.16
Conclusion
Understanding pathways of bone remodeling, including bone resorption and bone formation, is the key to future research and development of newer therapies for the prevention and treatment of osteoporosis. With the aging population in our country, the impact of menopause and the potential effects of certain medications, we are at risk for fragile bones and fracture. As rheumatologists, we need to continue to encourage nonmedical interventions for our younger population that can enhance their optimal peak bone mass. Additionally, there should be a major focus on nutrition, exercise and lifestyle modifications, all of which contribute to better bone health.
Ellen M. Field, MD, FACR, CCD, is a rheumatologist in private practice in Lehigh Valley, Pa.
References
- National Osteoporosis Foundation (NOF). www.nof.org.
- U.S. Department of Health and Human Services. Bone health and osteoporosis: A report of the Surgeon General. Rockville, Md.: U.S. Dept of Health and Human Services, Office of the Surgeon General; 2004. http://www.ncbi.nlm.nih.gov/books/NBK45513.
- Teitlebaum SL. Osteoclasts: What do they do and how do they do it? Am J Pathology. 2007;170(2):427–435. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1851862.
- Clemens TL, Rosen CJ. The insulin-like growth factor system and bone. Insulin-Like Growth Factors. Landes Bioscience. Austin, Texas, 2003.
- Kogianni, G, Noble BS. The biology of osteocytes. Curr Osteoporosis Rep. 2007;5(2):81–86.
- McClung MR, Lewiecki EM, Cohen SB, et al. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med. 2006;354:821–831.
- Bone HG, Bolognese MA, Yuen CK, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J Clin Endocrinol Metab. 2008;93(6):2149–2157.
- Cummings SR, San Martin J, McClung MR, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361(8):756–765.
- Brown JP, Prince RL, Deal C, et al. Comparison of the effect of denosumab and alendronate on BMD and biochemical markers of bone turnover in postmenopausal women with low bone mass: A randomized, blinded, phase 3 trial. J Bone Miner Res. 2009;24(1):153–161.
- Kendler DL, Roux C, Benhamou CL, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res. 2010;25(1):72–81.
- Logan Cy, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810.
- Ott SM. Sclerostin and Wnt signaling—The pathway to bone strength. J Clin Endocrinol Metab. 2005;90(12):6741–6743.
- Yadav VK, Ryu JH, Suda N, et al. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135(5):825–837.
- Goldring SR. Pathogenesis of bone and cartilage destruction in rheumatoid arthritis. Rheumatology. 2003;42(Suppl.2):ii11-ii16.
- Schett G, Hayer S, Zwerina J, et al. Mechanisms of disease: The link between RANKL and arthritic bone disease. Nat Clin Pract Rheumatol. 2005;1(1):47–54.
- Vuolteenaho K, Moilanen T, Hämäläinen M, et al. Effects of TNFα-antagonists on nitric oxide production in human cartilage. Osteoarthritis and Cartilage. 2002;10(4):327–332.