
Presentation
BF, a 43-year-old man with a history of attention deficit hyperactivity disorder (ADHD) and bipolar disorder, was in his usual state of excellent health until March 2014, when he experienced sudden onset of severe, sharp, left-sided abdominal pain radiating to the groin. The pain resolved after an hour, but recurred five times over the next week. Each episode was longer in duration, which prompted him to present to the emergency department of another hospital. He did not experience fevers, chills, night sweats, oral or genital ulceration, rash, visual changes, sinus symptoms, dyspnea, hematuria, or extremity weakness or numbness. His medications included lamotrigine without recent changes. His family and social histories were unremarkable.
On exam at the other hospital, he was afebrile and normotensive, with diffuse abdominal tenderness without guarding or rebound. Laboratory workup revealed a normal complete blood count, serum creatinine and urinalysis. Erythrocyte sedimentation rate (ESR) was 54 mm/hr (ref. <13 mm/hr) and
the C-reactive protein (CRP) was 5 mg/dL (ref. <0.5 mg/dL). A CT of the abdomen with oral and intravenous contrast was performed, which revealed a long left renal artery dissection with surrounding fat stranding, multiple left renal infarcts (see Figure 1) and dissections of both common and right external iliac arteries (see Figure 2). The right common iliac artery was noted to have surrounding soft tissue thickening. A CT angiogram of the chest did not demonstrate any abnormalities of the aorta or the great vessels.
In the absence of signs & symptoms suggestive of a specific etiology, the clinician must rely on inflammatory markers & imaging to make the diagnosis.
He was started on prednisone 40 mg twice daily and admitted to the other hospital. Further laboratory testing revealed negative anti-nuclear cytoplasmic antibody (ANCA), anti-nuclear antibody (ANA), cryoglobulins and a normal reaction to purified protein derivative (PPD). Complements, hepatitis B and C serologies, as well as a toxicology panel were also normal. His abdominal pain resolved during admission. A presumptive diagnosis of a large-vessel vasculitis was made.
Over the next month, he did not have recurrent abdominal pain and prednisone was decreased to 20 mg twice daily. A CT repeated six weeks after initial imaging revealed that the left common iliac dissection had extended into the aorta, with surrounding inflammatory changes. Fat stranding and soft tissue thickening surrounding the right common iliac and left renal artery had improved. His prednisone dose was increased to 60 mg daily, and he was referred to the rheumatology clinic at our hospital for evaluation.
On examination, he was afebrile, and his blood pressure was 145/85 mmHg with a heart rate of 121 beats per minute. He had easily visible veins on his shoulders, chest and abdomen but no hyperextensible joints. The remainder of the examination was unremarkable. Repeat laboratory testing revealed a thrombocytosis to 410 (ref. 150–400th/cu mm), ESR of 7 mm/hr (ref. <13 mm/hr) and a CRP of 35.2 mg/L (ref. <6 mg/L).
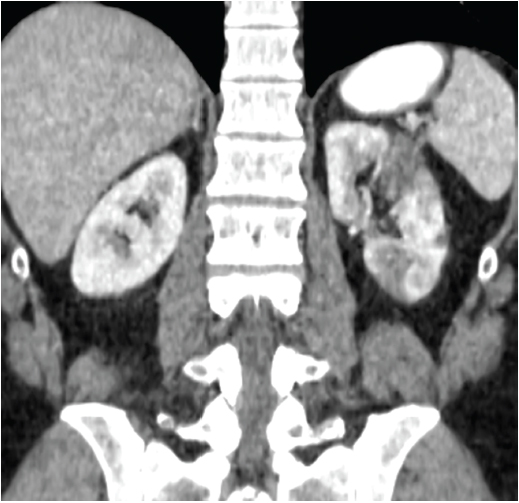
Note multiple hypodensities in the left renal cortex representing renal infarcts.
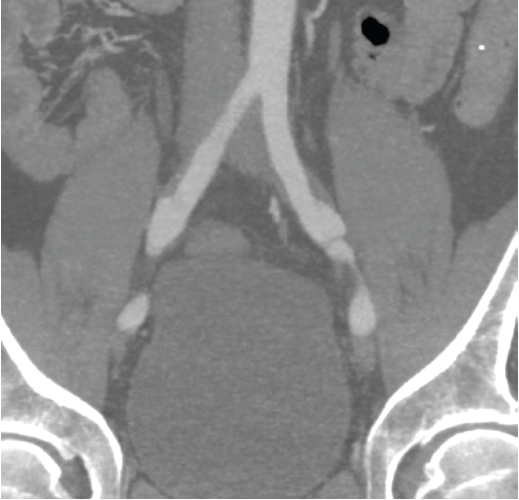
Note bilateral dissections of the common iliac arteries with surrounding soft tissue thickening.
Differential Diagnosis
We were faced with a patient who presented with multifocal dissections with associated inflammatory vessel wall changes as well as elevated inflammatory markers. Treatment with prednisone led to a decrease (although not complete resolution) of inflammatory marker elevations and resolution of some inflammatory changes; however, there was progression of the left common iliac artery dissection. A number of possible etiologies were considered, which can be broadly divided into vasculitides and noninflammatory vasculopathies.
Vasculitis
The most common vasculitis affecting the large vessels in an individual younger than 50 years is Takayasu arteritis (TAK). However, a number of other vasculitides, including Behçet’s and polyarteritis nodosa, can affect the large vessels, such as the renal and iliac arteries. Our patient did not have oral ulcerations to suggest Behçet’s or other signs or symptoms to suggest polyarteritis nodosa. Despite the elevated inflammatory markers, we felt that TAK was less likely because the inflammatory changes on imaging were limited to the areas of dissection and spared other vascular beds affected by TAK, particularly the aorta, subclavian and axillary arteries.1 Although TAK can lead to dissections, the most common arterial lesions are stenoses, which were not seen in our patient. In addition, 80–90% of patients with TAK are women, further decreasing the likelihood of this condition.1
Because our patient’s presentation was not consistent with a primary vasculitis, we felt that the elevated inflammatory markers and inflammatory changes surrounding the vessels on imaging were most likely secondary to the dissections, rather than primary manifestations of the underlying illness. These considerations raised our concern for a noninflammatory vasculopathy as the unifying diagnosis.
Noninflammatory Vasculopathies
Noninflammatory vasculopathies affecting the large vessels are a heterogeneous group of disorders and include Ehler’s Danlos Type IV (EDS IV), Marfan’s syndrome, Loeys-Dietz syndrome, fibromuscular dysplasia (FMD) and segmental arterial mediolysis (SAM). In addition, atherosclerotic lesions can lead to aneurysm and dissection and must be considered in the differential.
Marfan’s and Loeys-Dietz syndromes predominantly affect the aorta, which made these etiologies less likely in our patient. Although FMD preferentially affects the renal artery and can involve the iliacs, the typical “string of beads” appearance of the affected vessels, which is seen in approximately 90% of patients with FMD,2 was not seen on angiography, making this diagnosis less likely. In addition, FMD affects women in approximately 90% of cases, further arguing against this etiology.2 Our patient had no risk factors or imaging findings consistent with atherosclerotic disease. Therefore, we felt the most likely diagnoses were EDS IV or SAM.
EDS IV (vascular type) is a rare disorder that portends a significant risk of morbidity and mortality stemming from arterial or visceral rupture.3 Although EDS IV is inherited in an autosomal dominant pattern, studies suggest that approximately 50% of patients have de-novo mutations. In contrast to other types of EDS, large joint hypermobility is absent, although hyperextensibility of small joints can be observed in some patients. Other manifestations may include intestinal or uterine rupture, pneumothorax, translucent skin, characteristic facial appearance, acrogeria (aged appearance of the hands), tendon or muscle rupture, early onset of varicose veins and gingival recession.4 Eighty percent of patients are affected by the age of 40.3 Any large vessel can be affected in EDS IV, although the distal vessels of the extremities tend to be spared. The aorta, iliacs, renal, carotid and splanchnic vessels are most commonly affected.3 The diagnosis can be confirmed by testing for abnormalities in the COL3A1 gene, although some patients with EDS IV will not have detectable gene mutations.4
Our patient had a pattern of vascular involvement compatible with EDS IV, as well as easily visible veins, perhaps suggesting skin translucency. Therefore, we pursued testing for abnormalities in the COL3A1 gene.
SAM is a rare noninflammatory vasculopathy, which leads to vacuolization of tissue in the outer portion of the media and separation of the media from the adventitia (“mediolysis”), ultimately leading to aneurysm and/or dissection. SAM can affect individuals of any age, but generally affects patients in the fifth to sixth decade of life, with a slight male predominance.5 The renal arteries (sometimes leading to renal infarcts), celiac and superior mesenteric arteries and their branches are most commonly affected; however, iliac artery involvement has also been reported.5 Because of the arteries involved, histologic confirmation of the diagnosis can be challenging, and frequently, the diagnosis relies on imaging findings of dissection and/or aneurysm in the appropriate vascular distribution and exclusion of other etiologies discussed previously. Notably, in light of our patient’s presentation, several authors have reported that SAM can evolve rapidly, involving multiple vascular beds in a matter of weeks.6
Follow-Up
Prednisone was tapered off rapidly, and anticoagulation, blood pressure control and beta-blockade were started to prevent recurrence and progression of dissections. Over the next month, the patient’s inflammatory markers normalized. CT angiogram was repeated three months and nine months after initial presentation, demonstrating resolution of inflammatory changes and no new vascular lesions. Genetic testing for abnormalities in the COL3A1 gene was negative. Given the constellation of findings, we favored the diagnosis of SAM, although EDS type IV could not be completely ruled out.
Discussion
This case illustrates the diagnostic dilemma that rheumatologists confront when evaluating patients with large-vessel abnormalities. In the absence of signs and symptoms suggestive of a specific etiology (such as skin lesions and arthralgias seen in TAK, cranial symptoms seen in giant cell arteritis [GCA], oral and genital ulcers seen in Behçet’s, mononeuritis multiplex seen in polyarteritis nodosa or acrogeria and characteristic facies seen in EDS type IV), the clinician must rely on inflammatory markers and imaging to make the diagnosis.
Inflammatory markers can be helpful in the diagnostic evaluation; however, they have significant shortcomings. For example, in a study of 60 patients with TAK, only 72% had an elevated ESR during active disease, a finding consistent with other studies.7 In addition, although most patients with noninflammatory vasculopathy have normal inflammatory markers, the markers can be elevated in a minority of patients, particularly in the setting of acute dissection, as with our patient.
Given the limitations of laboratory testing, the interpretation of imaging studies is critical in differentiating between inflammatory and noninflammatory vascular processes. In our patient’s case, the presence of dissections without the long, smooth stenotic lesions seen in TAK and GCA, the absence of diffuse vascular involvement and the presence of inflammatory changes only in focal segments of dissection, all suggested a noninflammatory etiology. Further, the absence of lesions in the aorta and its branches, present in over 90% of patients with TAK and GCA, argued strongly against these etiologies.7
In summary, noninflammatory vasculopathies are important to consider in the evaluation of large-vessel abnormalities, (e.g., dissection, stenosis or aneurysm), renal infarcts and aortitis. A careful history, examination, review of imaging and laboratory testing (including genetic testing) are critical to making the correct diagnosis.
Eli Miloslavsky, MD, is an instructor in medicine at Harvard Medical School and Massachusetts General Hospital, Division of Rheumatology, Allergy and Immunology in Boston.
References
- Kerr GS. Takayasu’s arteritis. Rheum Dis Clin North Am. 1995;21:1041.
- Olin JW, Froehlich J, Gu X, et al. The United States Registry for Fibromuscular Dysplasia: Results in the first 447 patients. Circulation. 2012;125:3182.
- Oderich GS, Panneton JM, Bower TC, et al. The spectrum, management and clinical outcome of Ehlers-Danlos syndrome type IV: A 30-year experience. J Vasc Surg. 2005 Jul;42(1):98–106.
- Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000 Mar 9;342(10):673–680.
- Shenouda M, Riga C, Naji Y, Renton S. Segmental arterial mediolysis: A systematic review of 85 cases. Ann Vasc Surg. 2014 Jan;28(1):269–277.
- Michael M, Widmer U, Wildermuth S, et al. Segmental arterial mediolysis: CTA findings at presentation and follow-up. AJR Am J Roentgenol. 2006;187(6):1463–1469.
- Kerr GS, Hallahan CW, Giordano J, et al. Takayasu arteritis. Ann Intern Med. 1994; 120:919.


