Enabling early diagnosis & informed decision making
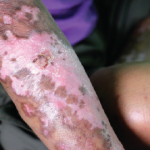
WASHINGTON, D.C.—Although most medical treatments are designed for the so-called average patient and use a one-size-fits-all approach, precision medicine seeks to tailor treatment to individuals based on their genes, environment, lifestyles and a host of other factors unique to them. At ACR Convergence 2024, the session titled, State of the Art Lecture: Precision Medicine in Systemic Sclerosis, brilliantly applied the concepts behind precision medicine to the care of patients suffering from scleroderma.
Very Early Diagnosis

Dr. Del Galdo
The first speaker was Francesco Del Galdo, MD, professor of experimental medicine (consultant), head of scleroderma program at the Leeds Institute of Rheumatic and Musculoskeletal Medicine, Leeds, U.K. Dr. Del Galdo is the current president of the European Scleroderma Trials and Research Group (EUSTAR) and, in this capacity, has been at the forefront of helping establish criteria for the very early diagnosis of systemic sclerosis (VEDOSS).