In individuals with SSc, PAH tends to have a poorer prognosis when compared with other World Health Organization Group I diagnoses. Recently, reports have demonstrated that survival in patients with isolated PAH receiving pulmonary vasodilator therapy is 78% at one year, 68% at two years, and 47% at three years.11,14,15 Unfortunately, survival of patients with respiratory-associated SSc-PAH is significantly worse than that of patients with isolated SSc-PAH.
Several groups have suggested exercise PAH may represent an intermediate SSc phenotype “between” normal and resting PH.16 In addition, Condliffe et al found 19% of SSc-PAH patients with exercise PH developed resting PAH after approximately 2.3 years, although controversy exists about the effect of comorbid conditions such as older age, persistent hypoxia, or systemic hypertension.14 Nevertheless, an evaluation of patients with SSc undergoing exercise may allow for earlier diagnosis and initiation of therapy, and, perhaps, a more favorable outcome of SSc-PAH.17
Several pathophysiologic mechanisms exist leading to elevated pulmonary arterial pressures in individuals with SSc. Because SSc is considered a subgroup belonging to the disease entity of PAH, the pathologic anatomy of pulmonary vessels has been regarded as similar to the vascular changes found in other forms of PAH.18 However, differences in clinical behavior and response to therapy between SSc-PAH and other forms of PAH are evident.19 This finding may be explained by coinciding patterns of arteriolar and a pulmonary venoocclusive disease–like pattern in the SSc population. Cohort and other studies continue to unravel these associations.
Gastrointestional Involvement
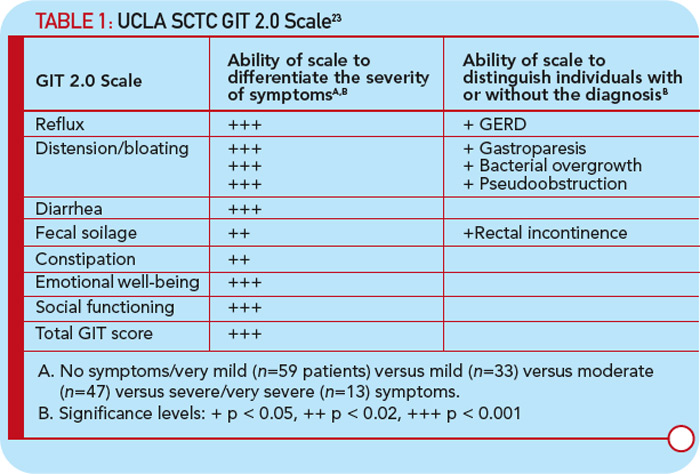
The gastrointestinal tract (GIT) is the most commonly involved internal organ system in SSc (approximately 90%), and gastroesophageal manifestations are frequent.20 As is true for SSc in general, there are two hypotheses for the pathogenesis of gastrointestinal (GI) involvement in SSc: vasculopathy and autoimmunity. Kawaguchi et al recently supported the autoimmune hypothesis by showing that within two years of onset of SSc, patients with severe GI involvement (malabsorption syndrome and/or pseudo-obstruction) had significantly higher titers and prevalence of anti–muscarinic-3 acetylcholine receptor antibody (anti-M3R) compared with patients with SSc without severe GI involvement.21