In 2009, two reports of genome-wide association studies (GWAS) in SSc, one involving American cases and the other involving European subjects, were presented at the annual meeting of the ACR in Philadelphia.3,4 Both studies found a strong association with the HLA Class II region on chromosome 6, firmly establishing that SSc is an autoimmune disease. The specific HLA Class II genes have been identified in a separate study.5 These were found to be different according to the SSc-specific autoantibody subgroup. In addition, several non-HLA candidate gene regions were found in the GWAS, but the exact identification of these genes has not yet been established.
There have been multiple reports of associations of non-HLA candidate genes with SSc, and the most likely candidates include those in pathways that regulate interferon production, such as interferon regulatory factor 5 (IRF5), as well as genes that regulate immunological responses such as signal transducer and activator 4 (STAT4). Gene variants in these pathways have been identified in systemic lupus erythematosus and in some populations of individuals with rheumatoid arthritis.6,7 This suggests that these genes may contribute to an increased susceptibility to autoimmunity in general. Future studies of candidate genes will identify functionally relevant disease pathways which may provide modifiable targets for therapy and provide a better classification schema with prognostic implications.
Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH) is a progressive disease that leads to increased pulmonary vascular resistance, compromised vasoreactivity, and eventual right-heart failure and death. This condition is an increasingly recognized cause of morbidity and mortality in SSc.8,9 Pulmonary hypertension (PH) is defined as a resting mean pulmonary arterial pressure >25mmHg at right-heart catheterization (RHC). PAH is defined as PH in addition to pulmonary capillary wedge <15mmHg.10 Based on RHC, the prevalence of resting PAH in SSc may be as high as 20%.11,12 However, histopathological evidence of pulmonary arteriopathy has been documented in up to 72% of patients with limited scleroderma, suggesting discordance between resting hemodynamics, clinical symptoms, and pathologic diagnosis.13
In individuals with SSc, PAH tends to have a poorer prognosis when compared with other World Health Organization Group I diagnoses. Recently, reports have demonstrated that survival in patients with isolated PAH receiving pulmonary vasodilator therapy is 78% at one year, 68% at two years, and 47% at three years.11,14,15 Unfortunately, survival of patients with respiratory-associated SSc-PAH is significantly worse than that of patients with isolated SSc-PAH.
Several groups have suggested exercise PAH may represent an intermediate SSc phenotype “between” normal and resting PH.16 In addition, Condliffe et al found 19% of SSc-PAH patients with exercise PH developed resting PAH after approximately 2.3 years, although controversy exists about the effect of comorbid conditions such as older age, persistent hypoxia, or systemic hypertension.14 Nevertheless, an evaluation of patients with SSc undergoing exercise may allow for earlier diagnosis and initiation of therapy, and, perhaps, a more favorable outcome of SSc-PAH.17
Several pathophysiologic mechanisms exist leading to elevated pulmonary arterial pressures in individuals with SSc. Because SSc is considered a subgroup belonging to the disease entity of PAH, the pathologic anatomy of pulmonary vessels has been regarded as similar to the vascular changes found in other forms of PAH.18 However, differences in clinical behavior and response to therapy between SSc-PAH and other forms of PAH are evident.19 This finding may be explained by coinciding patterns of arteriolar and a pulmonary venoocclusive disease–like pattern in the SSc population. Cohort and other studies continue to unravel these associations.
Gastrointestional Involvement
The gastrointestinal tract (GIT) is the most commonly involved internal organ system in SSc (approximately 90%), and gastroesophageal manifestations are frequent.20 As is true for SSc in general, there are two hypotheses for the pathogenesis of gastrointestinal (GI) involvement in SSc: vasculopathy and autoimmunity. Kawaguchi et al recently supported the autoimmune hypothesis by showing that within two years of onset of SSc, patients with severe GI involvement (malabsorption syndrome and/or pseudo-obstruction) had significantly higher titers and prevalence of anti–muscarinic-3 acetylcholine receptor antibody (anti-M3R) compared with patients with SSc without severe GI involvement.21
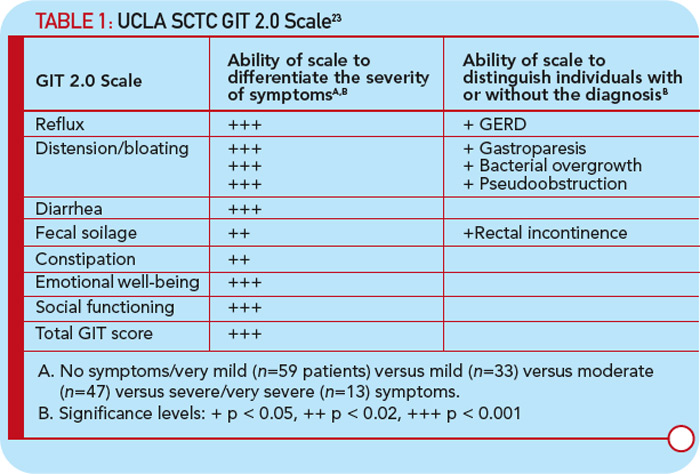
Baron et al investigated 586 patients from the Canadian Scleroderma Research Group Registry and used a validated instrument to assess malnutrition: the malnutrition universal screening tool.22 These investigators showed that 18% of their cohort were at high risk for malnutrition and that malnutrition was associated with increased disease severity, shorter disease duration, and more frequent GIT involvement. Large randomized controlled trials have not been conducted in individuals with GIT involvement, partly due to lack of a validated instrument. Khanna is leading a team of researchers at three large scleroderma centers to develop a fully validated GI instrument, the University of California, Los Angeles (UCLA) Scleroderma Clinical Trial Consortium (SCTC) GIT 2.0. This instrument includes 34 items with seven scales: reflux, distention/bloating, diarrhea, fecal soilage, constipation, emotional well-being, and social functioning.23 Table 1 (below) depicts symptom severity and disease correlations supporting the reliability and validity of the method. This instrument is currently being used as a secondary outcome measure in several ongoing randomized clinical trials in the treatment of SSc.
Therapy
In double-blind trials, treatment with methotrexate was shown to be effective for the skin involvement but with moderate effect; it may potentially have an effect on the lungs, although this is not proven statistically. A large randomized, placebo-controlled trial of cyclophosphamide suggested a small effect in reducing loss for forced vital capacity (FVC) while improving measures of quality of life and dyspnea.24 Subsequent exploratory analysis suggested that the magnitude of the effect was strongly related to disease severity at treatment outset, includingthe level of FVC and extent of fibrosis by high-resolution computerized tomography (HRCT) of the lung. “Traditional” measures of activity (inflammation) including cellularity on bronchoalveolar lavage and “ground glass” on HRCT predicted neither the clinical course nor the response to therapy. Other prospective data sets offered complementary information.25 The implications are important for bedside decision making and for design of future trials.
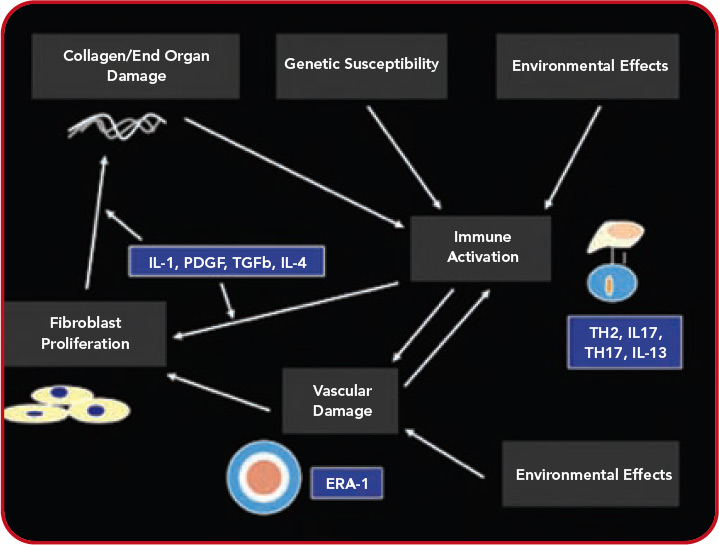
Conventional treatments are directed nonspecifically as macrophage/lymphocyte activity therapy (methotrexate) or as nonspecific alkylating therapy (cyclophosphamide). On the other hand, with increasing understanding of the pathogenesis of SSc, the possibility of more targeted therapy is beginning to evolve. As shown in Figure 1 (above), a number of cytokines and proteins have potential roles in the pathogenesis of this disease. Anti–TGF-beta was tested in a pilot study of SSc, but was not effective, although the pilot nature of this trial makes it very underpowered.26 This substance or congeners may still find a place in the therapy of SSc. Suppression of PDGF through combined c-Abl/PDGF TK receptor binders such as imatinib, nilotinib, or dasatinib has undergone a few open label studies and possible encouraging results will be tested in well-controlled trials.27 Because endothelin receptor antagonists have an antifibrotic effect in animals, they have also been tested in patients with SSc. The results of these studies, however, have not been fully published, although data regarding use of endothelin receptor antagonists in PAH and bosentan in digital ulcers indicates the possibility that this class of drugs has a vascular effect.28
Over the last year, there has been increasing interest in other targets displayed in Figure 1 (above). For example, interleukin (IL) 4, which is increased in the bronchoalveolar lavage fluid, blood, and skin of patients with SSc might be considered as a possible target for therapy.29 Radstake et al point out the possible role of IL1, IL17, and Th17 cells in this disease.30 In addition, CTGF presents a possible target for systemic sclerosis.31 While definitive therapy is not yet at hand, the past year indicates the possibility that targeted therapy may assume a role in at least some aspects of SSc.
Stem Cell Transplantation
As described above, progress continues in our understanding of the biology and pathogenesis of SSc. Similarly, progress has been observed in the application of hematopoietic cell transplantation (HCT) for the treatment of severe autoimmune diseases. In the 1980s, animal experiments in genetic or antigen-induced models of autoimmune disorders demonstrated that following myeloablative conditioning, allogeneic or autologous HCT can prevent disease progression and even reverse organ damage in, respectively, inherited or acquired models of autoimmune disease.32,33
Remarkably, studies of post-transplant immune recovery showed that the reconstituted immune repertoire following lymphoablation and autologous HCT demonstrated repair of baseline immune defects and alterations in adoptive immune responses associated with the autoimmune disease.34 Following allogeneic HCT, a normal donor immune system is engrafted in the transplant recipient. With CD34+ selected autologous HCT, the immune network is normalized and “reset” via repertoire replacement.34 At present, studies are ongoing to determine whether and to what extent suppression of inflammation after HCT is due to re-regulation of immunity or abolition of populations of disease-associated T or B cells.

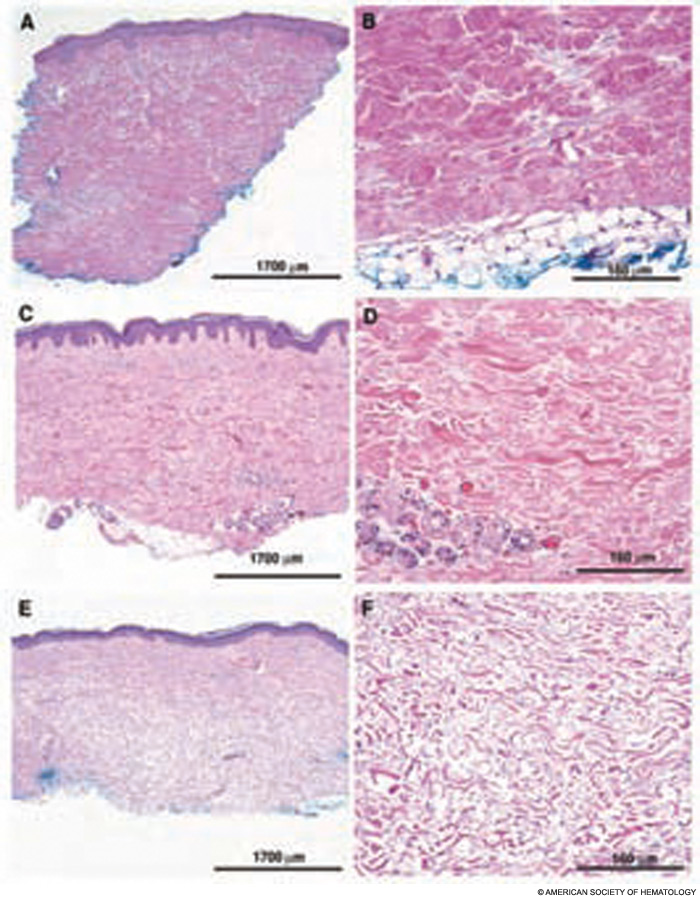
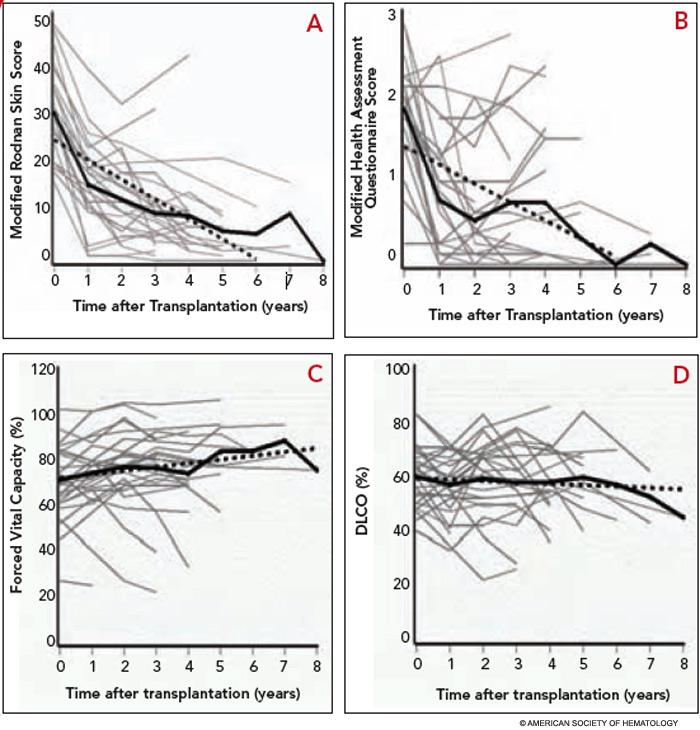
Completed clinical pilot studies provide data with sufficient follow-up to weigh the efficacy and safety of HCT for autoimmune diseases.35 More than 1,000 transplants for autoimmune diseases have been performed in Europe. Ongoing phase III multinational European trials are being conducted in SSc (the ASTIS trial), multiple sclerosis (the ASTIMS study), and Crohn’s disease (the ASTIC trial). Completed phase II trials in the United States of HCT for SSc have shown promising outcomes. Figures 2 (p. 21) and 3 (p. 22) demonstrate significant improvement/resolution of dermal sclerosis and stabilization/improvement in pulmonary function persisting beyond five years after autologous HCT.36 A follow-up study is comparing treatment with chronic high-dose pulse immunosuppression versus lymphoablation and autologous CD34+ selected autologous HCT. This phase III randomized study is currently recruiting patients in Canada and the United States who have severe SSc and internal organ involvement for enrollment in the SCOT (Scleroderma: Cyclophosphamide or Transplantation) trial. Most commonly, individuals with diffuse scleroderma are enrolled who have baseline FVC or DLCO values <70% predicted. Table 2 (p. 22) provides an overview of the eligibility and referral information with further details found at the study Web site (www.sclerodermatrial.org). Within the trial are imbedded a number of NIH-supported mechanistic studies to further understand the molecular mechanisms of SSc, immune regulation, and response to treatment.
Conclusions
Important advances have been gained in our understanding of the genetics, pathobiology and treatment of severe SSc. While SSc is often said to be the most fearsome of the autoimmune diseases, molecular targets for therapy have been defined and pilot trials of targeted therapies are underway. Convincing animal and human data demonstrate that the immune repertoire can be reset after hematopoietic stem cell transplantation leading to dramatic reversal of dermal fibrosis and stabilization to improvement of pulmonary disease. A pivotal National Institutes of Health–sponsored randomized trial comparing immunosuppression to immunoablative HCT is currently underway and patients are being recruited to the SCOT trial to test outcomes in these two promising treatments. Such trials will provide basic scientists with a better understanding of the regulatory pathways of SSc and rheumatologists with a context to weigh progress in the treatment of scleroderma.
Acknowledgments
This work was supported in part by NIH award AI-05419. The authors have nothing else to disclose.
Dr. Furst is professor in the division of rheumatology at UCLA; Dr. Mayes is professor in the division of rheumatology at the University of Texas in Houston; Dr. Khanna is assistant professor of rheumatology at UCLA; Dr. Seibold is professor of rheumatology at the University of Connecticut in Farmington; Dr. Saggar a clinical instructor in the division of pulmonary and critical care at UCLA; and Dr. Sullivan is the James B. Wyngaarden Professor of Medicine in the division of cellular therapy at Duke University Medical Center in Durham, N.C.
References
- Arnett FC, Cho M, Chatterjee S, Aguilar MB, Reveille JD, Mayes, MD. Familial occurrence frequencies and relative risks for systemic sclerosis (scleroderma) in three US cohorts. Arthritis Rheum. 2001; 44:1359-1362.
- Assassi S, Arnett FC, Reveille JC, Gourh P, Mayes MD. Clinical, immunological, and genetic features of familial systemic sclerosis. Arthritis Rheum. 2007: 56:2031-2037.
- Gorlova O, Weng S-F, Ying J, et al. Genome-wide association study of systemic sclerosis in a large U.S. cohort of over 1,500 cases. Arthritis Rheum. 2009; 60:S203 (abstract).
- Radstake TRDJ, Alizadeh BZ, Palomino-Morales R, et al. Genome-wide association scan in systemic sclerosis identified MHC region and two additional susceptibility loci on 2q32 and 7q32. Arthritis Rheum. 2009; 60:S473 (abstract).
- Arnett FC, Gourh P, Shete S, et al. Major Histocompatibility Complex (MHC) class II alleles, haplotypes, and epitopes which confer susceptibility or protection in the fibrosing autoimmune disease systemic sclerosis: analyses in 1300 Caucasian, African-American and Hispanic cases and 1000 controls. Ann Rheum Dis. 2009; Jul 12 (epub ahead of print).
- Scofield RH. Genetics of systemic lupus erythematosus and Sjogrens syndrome. Curr Opin Rheumatol. 2009; 21:448-453.
- Ji JD, Lee WJ, Kong KA, et al. Association of STAT4 polymorphism with rheumatoid arthritis and systemic lupus erythematosus: a meta-analysis. Mol Biol Rep. 2010; 37:141-147.
- Badesch DB, Abman SH, Simonneau G, Rubin LJ, McLaughlin VV. Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest. 2007; 131:1917-1928.
- Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:55-125.
- Badesch DB, Champion HC, Sanchez MA, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009; 54:555-566.
- Mukerjee D, St George D, Coleiro B, et al. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: application of a registry approach. Ann Rheum Dis. 2003;62:1088-1093.
- Ungerer RG, Tashkin DP, Furst D, et al. Prevalence and clinical correlates of pulmonary arterial hypertension in progressive systemic sclerosis. Am J Med. 1983; 75:65-74.
- D’Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med. 1969;46:428-440.
- Condliffe R, Kiely DG, Peacock AJ, et al. Connective tissue disease-associated pulmonary arterial hypertension in the modern treatment era. Am J Respir Crit Care Med. 2009;179:151-157.
- Mathai SC, Hummers LK, Champion HC, et al. Survival in pulmonary hypertension associated with the scleroderma spectrum of diseases: impact of interstitial lung disease. Arthritis Rheum. 2009;60:569-577.
- Tolle JJ, Waxman AB, Van Horn TL, Pappagianopoulos PP, Systrom DM. Exercise-induced pulmonary arterial hypertension. Circulation. 2008;118:2183-2189.
- Proudman SM, Stevens WM, Sahhar J, Celermajer D. Pulmonary arterial hypertension in systemic sclerosis: the need for early detection and treatment. Intern Med J. 2007;37:485-494.
- Cool CD, Kennedy D, Voelkel NF, Tuder RM. Pathogenesis and evolution of plexiform lesions in pulmonary hypertension associated with scleroderma and human immunodeficiency virus infection. Hum Pathol. 1997;28:434-442.
- Fisher MR, Mathai SC, Champion HC, et al. Clinical differences between idiopathic and scleroderma-related pulmonary hypertension. Arthritis Rheum. 2006;54:3043-3050.
- Sjogren RW. Gastrointestinal features of scleroderma. Curr Opin Rheumatol. 1996; 8(6):569-575.
- Kawaguchi Y, Nakamura Y, Matsumoto I, et al. Muscarinic-3 acetylcholine receptor autoantibody in patients with systemic sclerosis: contribution to severe gastrointestinal tract dysmotility. Ann Rheum Dis. 2009;68:710-714.
- Baron M, Hudson M, Steele R. Malnutrition is common in systemic sclerosis: results from the Canadian scleroderma research group database. J Rheumatol. 2009;36:2737-2743.
- Khanna D, Hays RD, Maranian P, et al. Reliability and validity of the University of California, Los Angeles Scleroderma Clinical Trial Consortium Gastrointestinal Tract Instrument. Arthritis Rheum. 2009;61:1257-1263.
- Tashkin DP, Elashoff R, Clements PJ, et al. Effects of a one-year treatment with cyclophosphamide on outcomes at two years in scleroderma lung disease. Am J Resp Crit Care Med. 2007;176:1026-1034.
- Goh, N, Sujal SL, Desai R, et al. Interstitial lung disease in systemic sclerosis: A simple staging system. Am J Respir Crit Care Med. 2008;177:1248-1254.
- Denton CP, Merkel PA, Furst DE, et al. Recominant human anti-transforming growth factor beta 1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007;56:323-333.
- Distler JH, Distler O. Intracellular tyrosine kinases as novel targets for anti-fibrotic therapy in systemic sclerosis. Rheumatology (Oxford). 2008; 47(Suppl 5):v10-v11.
- Korn JH, Mayes M, Matucci Cerinic M, et al. Digital ulcers in systemic sclerosis: prevention by treatment with bosentan, an oral endothelin receptor antagonist. Arthritis Rheum. 2004;50:3985-3993.
- Atamas SP, White B. The role of chemokines in the pathogenesis of scleroderma. Curr Opin Rheumatol. 2003;15:772-777.
- Radstake TR, van Bon L, Broen J, et al. The pronounced Th17 profile in systemic sclerosis (SSc) together with intracellular expression of TGFbeta and IFNgamma distinguishes SSc phenotypes. PLoS One. 2009;4(6):e5903.
- Della Bella S, Molteni M, Mascagni B, et al. Cytokine production in scleroderma patients: effects of therapy with either iloprost or nifedipine. Clin Exp Rheumatol. 1997; 15:135-141.
- Ikehara S, Good Ra, Nakamura T, et al. Rationale for bone marrow transplantation in the treatment of autoimmune diseases. Proc Natl Acad Sci USA. 1985;82:2483-2487.
- van Bekkum DW, Bohre EP, Houben PF, Knaan-Shanzer S. Regression of adjuvant-induced arthritis in rats following bone marrow transplantation. Proc Natl Acad Sci USA. 1989;86:1090-1094.
- Muraro PA, Douek DC, Packer A, et al. Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J Exp Med. 2005;201:805-816.
- Sullivan KM, Muraro P, Tyndall A. Hematopoietic cell transplantation for autoimmune disease: updates from Europe and the United States. Biol Blood Marrow Transplant. 2010;16:S48-S56.
- Nash RA, McSweeney PA, Crofford LJ, et al. High-dose immunosuppressive therapy and autologous hematopoietic cell transplantation for severe systemic sclerosis: long-term follow-up of the US multicenter pilot study. Blood. 2007;110:1388-1396.