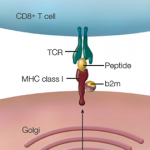
Adaptive immunity was now an established and experimentally testable construct. But what if the self/nonself discrimination was flawed? Would that not set the stage of a “horror autotoxicus” that would unleash the power of the immune system on one’s own tissue?
Rebecca Lancefield, PhD, working in New York, had specialized in immunochemical studies of group A streptococci. In 1928, she published a study on the chemical composition and antigenicity of streptococci. In 1933, she introduced a new method of classifying streptococci into groups according to the ability of antigens in their cell walls to induce the formation of precipitating antibodies. These groups came to known as Lancefield groups. In 1946, Maclyn McCarty, MD, was asked to head the laboratory at Rockefeller University in New York that had been established to work on streptococci and rheumatic fever. Clinical observations, combined with Dr. Lancefield’s classification, had established that acute rheumatic fever was a late complication of group A streptococcal pharyngitis. Dr. McCarty approached this problem by studying both the biology of group A streptococci and patients with acute rheumatic fever admitted to the Rockefeller Hospital.
In the fertile culture of Rockefeller University on streptococcal research, the team began to examine the appearance of heart-reactive antibodies that could be detected in the serum of patients with rheumatic fever. John Zabriskie, MD, effectively took a page from Burnet’s observations and asked whether the appearance of heart-reactive antibodies could actually represent, not a failure of self/nonself discrimination, but rather the misfortune of antigenic cross-reactivity between streptococcal determinants and cardiac proteins.1 Absorption of heart-reactive antibodies by streptococcal cell walls supported this notion, and the theory of molecular mimicry began to gain experimental support.2 This particular cross-reactivity was subsequently shown to relate to the M protein of the streptococcal cell wall and cardiac myosin.3 Yet, for all its elegance in linking infection to autoimmunity, molecular mimicry remained difficult to prove rigorously.4
Hematologists in search of the cause of pernicious anemia might have probed the bone marrow ad infinitum without realizing that the answer lay in the gut. Are rheumatologists destined to learn the same lesson?
Reactive Arthritis Reconsidered
Reactive arthritis (ReA) became operationally defined as a nonseptic arthritis that followed an extraarticular infection. Although rheumatic fever itself would qualify for this definition, the concept became clinically associated with antecedent infections of the gastrointestinal (GI) and genitourinary (GU) tracts. In the case of Salmonella or Chlamydia cultured from the respective sites, this construct provided a useful framework to understanding pathogenesis. However, much of the clinical research in this area was hampered by uncertainties in disease definition. If the triggering pathogen could not be isolated from the GI or GU tract, would antecedent diarrhea or urethritis be sufficient for the diagnosis? Would there be antibody profiles with sufficient specificity to be considered as inclusion criteria for ReA? Would proliferation of immunoreactive cells from synovial fluid (in response to candidate bacterial antigens) carry diagnostic specificity? Challenges to these studies were also compounded by poorly defined diagnostic criteria for ReA, and by changing nomenclature, as the older term Reiter’s syndrome came to be replaced by reactive arthritis.