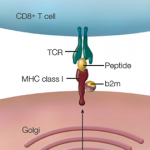
When seen in the context of concurrent genetic polymorphisms, not only in TLR but also in class I MHC and IL23-R, the infectious link between gut and joint has become intriguingly complex. This situation has become clinically relevant with the recognition that TLR2 genetic polymorphisms predispose to post-Salmonella ReA. The importance of the gut as the site of triggering events has been supported not only by the recognition that the same genetic variants confer susceptibility to AS and to Crohn’s disease (CD) but also that subclinical inflammation and upregulation of IL-23 expression is present in gut tissues of AS patients.15,16 Hematologists in search of the cause of pernicious anemia might have probed the bone marrow ad infinitum without realizing that the answer lay in the gut. Are rheumatologists destined to learn the same lesson?
Too Much—or Too Little?—of a Good Thing
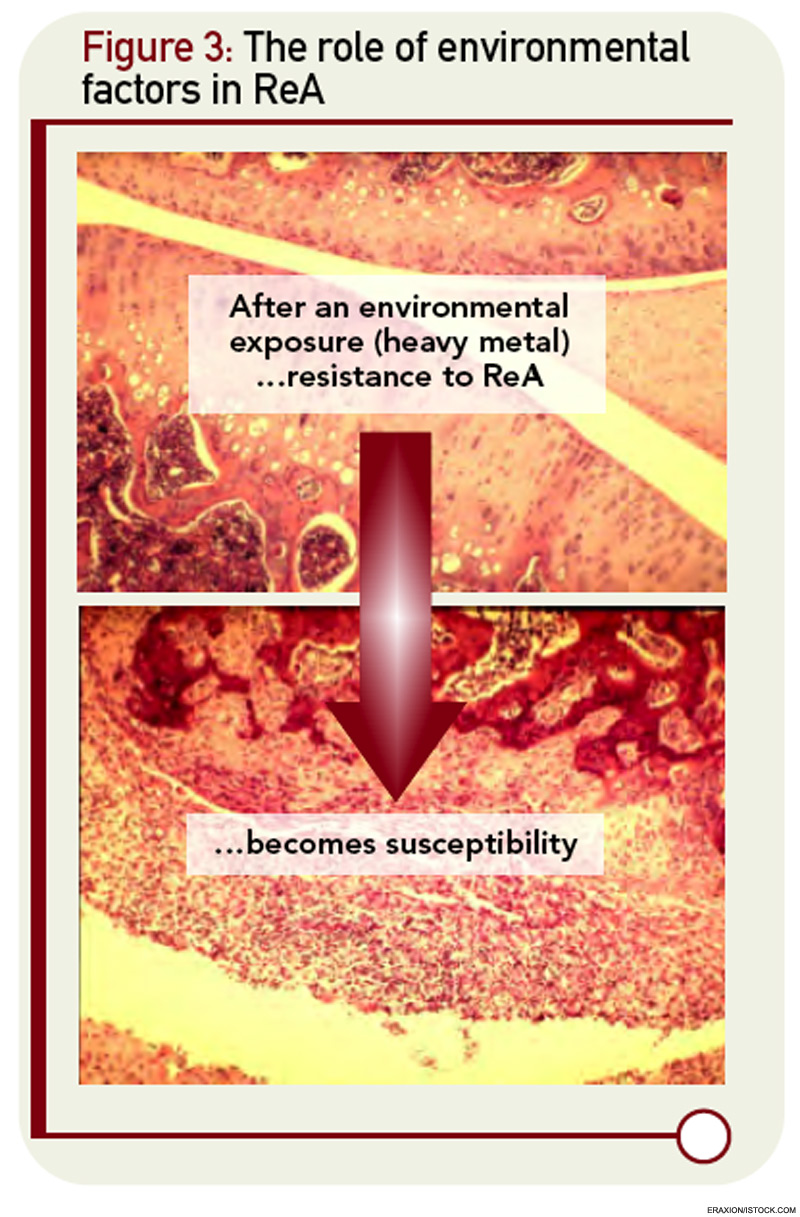
The underlying assumption in the molecular mimicry theories, exemplified by the studies of rheumatic fever, is that the host immune response to infection is overactive in nonseptic sequelae like ReA. But there are intriguing clues that host defenses might be diminished rather than amplified in patients with ReA. The cytokine profiles of ReA patients suggest down-regulation of proinflammatory cytokines rather than upregulation.17 These changes include a decrease in levels of TNF-α and IFN-γ, cytokines normally thought to be deleterious rather than beneficial cytokines with respect to arthritis. The role of cytokine dysregulation in disease etiology is borne out in experimental ReA.18 Animals that are susceptible to Chlamydia-induced arthritis have diminished rather than enhanced production of TNF-α and IFN-γ within the joint. This appears to relate to impaired capacity of host clearance of the organism in susceptible strains of animals. Of interest, this genetically defined cytokine signature can be dramatically altered by exposure to heavy metals, suggesting that other environmental factors may come into play in the dynamics of ReA in the clinical setting (see Figure 3, below right).19