At the same time, Ilya Metchnikoff had established a private laboratory at Messina, Italy, to study comparative embryology. There he discovered the phenomenon of phagocytosis after experimenting on the larvae of starfish. His theory—that specific white blood cells could engulf and destroy harmful pathogens such as bacteria—was met with skepticism from leading specialists, including Pasteur. Nonetheless, the conceptual door was opened to recognizing that the outcome of infection was not solely the domain of the invader, but very much dependent on the strength of host defenses, epitomized by the macrophage. The battle lines were drawn for defence of the host. For an astute observer of disease like Osler, it was not a stretch to imagine that arthritis, like meningitis or cellulitis, would turn out to be infectious in origin.
Self, Streptococcus, and Immunity
In 1949, Sir Frank Burnet borrowing a concept well established in psychology by Freud and Jung, first used the term “self” in the context of immunology. With this foundational principle, Burnet proposed in 1955 that the fundamental role of the immune system is to discriminate between self and nonself. With the clonal selection theory, he operationalized this concept to argue that protein determinants, which are regarded as foreign by the immune system, can lead to an expansion of immunocompetent cells with specificity for the antigen. Burnet was awarded the Nobel Prize in Medicine and Physiology in 1960.
Adaptive immunity was now an established and experimentally testable construct. But what if the self/nonself discrimination was flawed? Would that not set the stage of a “horror autotoxicus” that would unleash the power of the immune system on one’s own tissue?
Rebecca Lancefield, PhD, working in New York, had specialized in immunochemical studies of group A streptococci. In 1928, she published a study on the chemical composition and antigenicity of streptococci. In 1933, she introduced a new method of classifying streptococci into groups according to the ability of antigens in their cell walls to induce the formation of precipitating antibodies. These groups came to known as Lancefield groups. In 1946, Maclyn McCarty, MD, was asked to head the laboratory at Rockefeller University in New York that had been established to work on streptococci and rheumatic fever. Clinical observations, combined with Dr. Lancefield’s classification, had established that acute rheumatic fever was a late complication of group A streptococcal pharyngitis. Dr. McCarty approached this problem by studying both the biology of group A streptococci and patients with acute rheumatic fever admitted to the Rockefeller Hospital.
In the fertile culture of Rockefeller University on streptococcal research, the team began to examine the appearance of heart-reactive antibodies that could be detected in the serum of patients with rheumatic fever. John Zabriskie, MD, effectively took a page from Burnet’s observations and asked whether the appearance of heart-reactive antibodies could actually represent, not a failure of self/nonself discrimination, but rather the misfortune of antigenic cross-reactivity between streptococcal determinants and cardiac proteins.1 Absorption of heart-reactive antibodies by streptococcal cell walls supported this notion, and the theory of molecular mimicry began to gain experimental support.2 This particular cross-reactivity was subsequently shown to relate to the M protein of the streptococcal cell wall and cardiac myosin.3 Yet, for all its elegance in linking infection to autoimmunity, molecular mimicry remained difficult to prove rigorously.4
Hematologists in search of the cause of pernicious anemia might have probed the bone marrow ad infinitum without realizing that the answer lay in the gut. Are rheumatologists destined to learn the same lesson?
Reactive Arthritis Reconsidered
Reactive arthritis (ReA) became operationally defined as a nonseptic arthritis that followed an extraarticular infection. Although rheumatic fever itself would qualify for this definition, the concept became clinically associated with antecedent infections of the gastrointestinal (GI) and genitourinary (GU) tracts. In the case of Salmonella or Chlamydia cultured from the respective sites, this construct provided a useful framework to understanding pathogenesis. However, much of the clinical research in this area was hampered by uncertainties in disease definition. If the triggering pathogen could not be isolated from the GI or GU tract, would antecedent diarrhea or urethritis be sufficient for the diagnosis? Would there be antibody profiles with sufficient specificity to be considered as inclusion criteria for ReA? Would proliferation of immunoreactive cells from synovial fluid (in response to candidate bacterial antigens) carry diagnostic specificity? Challenges to these studies were also compounded by poorly defined diagnostic criteria for ReA, and by changing nomenclature, as the older term Reiter’s syndrome came to be replaced by reactive arthritis.
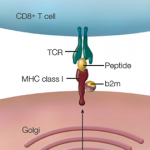
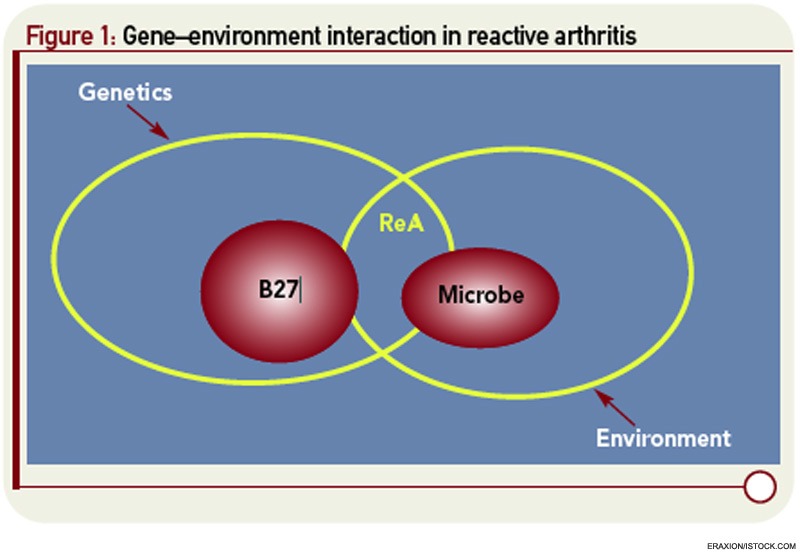
Extrapolating from rheumatic fever, a number of investigations addressed molecular mimicry as the basis of ReA. These studies sought to operationalize the recent discovery of HLA-B27 as a risk factor not only for ReA but also for ankylosing spondylitis (AS). Approaches to linking infection to disease included demonstration of sequence homology between B27 and arthritogenic pathogens, as well as the characterization of monoclonal antibodies and T-cell clones reacting with both B27 and the putative inciting organism (see Figure 1, p. 24).6,7 Some experimental models suggested that Chlamydia could break tolerance of self-B27.8 However, rigorous proof that autoreactivity is the underlying mechanism in ReA remained difficult to obtain, and ReA remained a charter member of the seronegative spondyloarthropathies.
The report that phlogistic components of Yersinia and Salmonella could be recovered from synovial fluid of patients with ReA suggested that, rather than autoimmunity acting as the operational event in chronic ReA, local persistence of microbial fragments could be the true culprit. The presence of such foreign material was demonstrated by immunofluorescence studies of synovial fluid and synovial tissues.9 These findings shifted the focus of investigators to address the structural basis of arthritogenicity. Thus, certain glycosylation patterns of foreign antigens appeared to correspond with the ability to induce experimental ReA, by virtue of lingering as nonbiodegradable fragments in the joint.10 Gas chromatography–mass spectrometry analysis of organisms could identify such patterns, but this approach has yet to be transferred into clinical practice.
Innate Immunity Weighs In
For all the sophistication of adaptive immunity, with specificity and memory as its pinnacle achievements, it was recognized that the time required to expand the T-cell and B-cell responses in the setting of acute infection could place the host in great peril. The concept of innate immunity as the first line of host defense had been the province of leukocyte biology students, subsequently drosophila scholars. The discovery of toll-like receptors (TLRs) as the quintessential recognition elements of the innate immune system created an intense interest in defining the role of this family of receptors in infectious disease. However, the initial concepts proposed by the late Charles Janeway, MD, and colleagues seemed to have placed some conceptual hurdles in the path of linking TLR to rheumatic diseases.
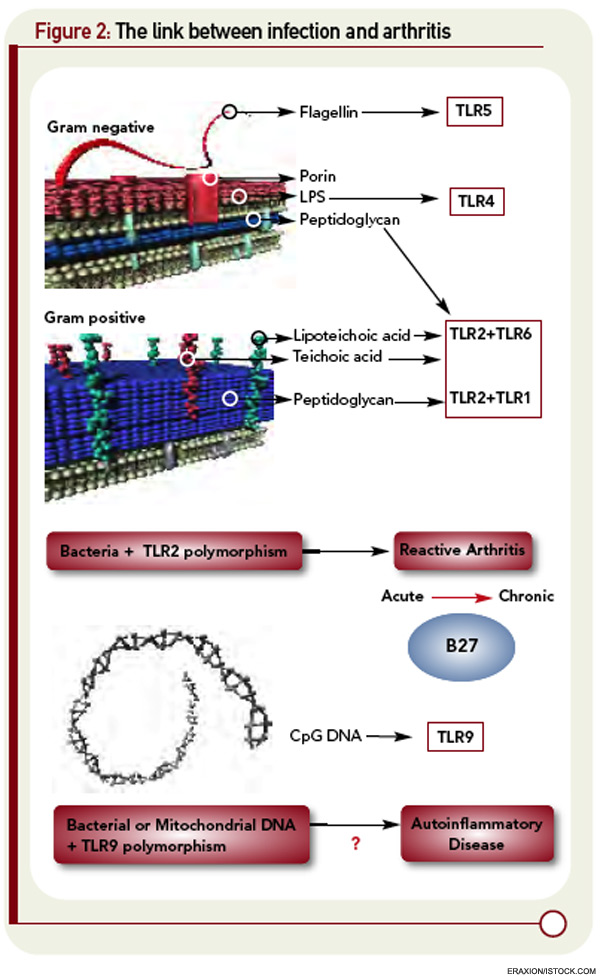
The first of these hurdles related to the apparent nonpolymorphic nature of the TLR, posing a significant challenge to addressing the heterogeneity of susceptibility to ReA in target populations. The second was that self/nonself discrimination by the innate immune system was proposed to be perfect, unlike its forever failing counterpart in adaptive immunity. Both of these concepts have needed significant revision. There are, in fact, significant polymorphisms in the TLR families, and recent studies in epidemic ReA have demonstrated that nonsynonymous single nucleotide polymorphisms in TLR2 confer susceptibility to post-Salmonella ReA (see Figure 2, p. 28).11 The notion that TLRs are specific only for foreign determinants has been replaced with the recognition that mammalian nucleic acids, among other molecules, are ligands for certain TLRs; indeed, this concept has proved to be pivotal in current concepts of lupus pathogenesis.
These findings have broadened the landscape linking infection to arthritis. Microbial pathogenesis began from the conceptual starting line that disease mediated by infection resulted from viable microbes in the target organ, as a logical application of Dr. Koch’s postulates. The discovery of the complex tango of receptor–ligand interactions involving TLRs led to the recognition that the inflammation arising as a consequence of infection requires neither a viable pathogen nor even of the intact organism. Signaling events that could usher in damaging inflammation called only for fragments of the pathogen, such as LPS or peptidoglycan, to interact with TLRs on host cells. This has been borne out in several experimental models of arthritis. The B27 transgenic rat expresses a number of features which recapitulate spondyloarthropathy (SpA) in patients, including arthritis, dermatitis, and enteritis.12 These features are dramatically attenuated if the animal is raised in a germ-free environment.13 Re-establishing the clinical features of SpA in this model does not require a gut infection in the usual sense. Rather, the animal need only be challenged with a gut commensal, Bacteroides, which would normally not be counted among the arthritogenic pathogens.
Thus, the gut being populated, not infected, with commensal organisms creates the immunological platform on which arthritis can proceed. In the postcolonial era of the twenty-first century, most find the notion of being perpetually colonized somewhat disconcerting. The interleukin (IL)-1Ra–deficient mouse is genetically programmed to develop an aggressive polyarthritis. This propensity also is completely abrogated if the animal is raised germ free.14 To re-establish the arthritis in the germ-free setting requires no intact organism at all. A TLR ligand alone will do quite nicely. These studies also have demonstrated that TLR signaling at the gut level can have very disparate effects on arthritis, with TLR4 signaling playing an exacerbating role and TLR2 signaling playing a modulatory role.
When seen in the context of concurrent genetic polymorphisms, not only in TLR but also in class I MHC and IL23-R, the infectious link between gut and joint has become intriguingly complex. This situation has become clinically relevant with the recognition that TLR2 genetic polymorphisms predispose to post-Salmonella ReA. The importance of the gut as the site of triggering events has been supported not only by the recognition that the same genetic variants confer susceptibility to AS and to Crohn’s disease (CD) but also that subclinical inflammation and upregulation of IL-23 expression is present in gut tissues of AS patients.15,16 Hematologists in search of the cause of pernicious anemia might have probed the bone marrow ad infinitum without realizing that the answer lay in the gut. Are rheumatologists destined to learn the same lesson?
Too Much—or Too Little?—of a Good Thing
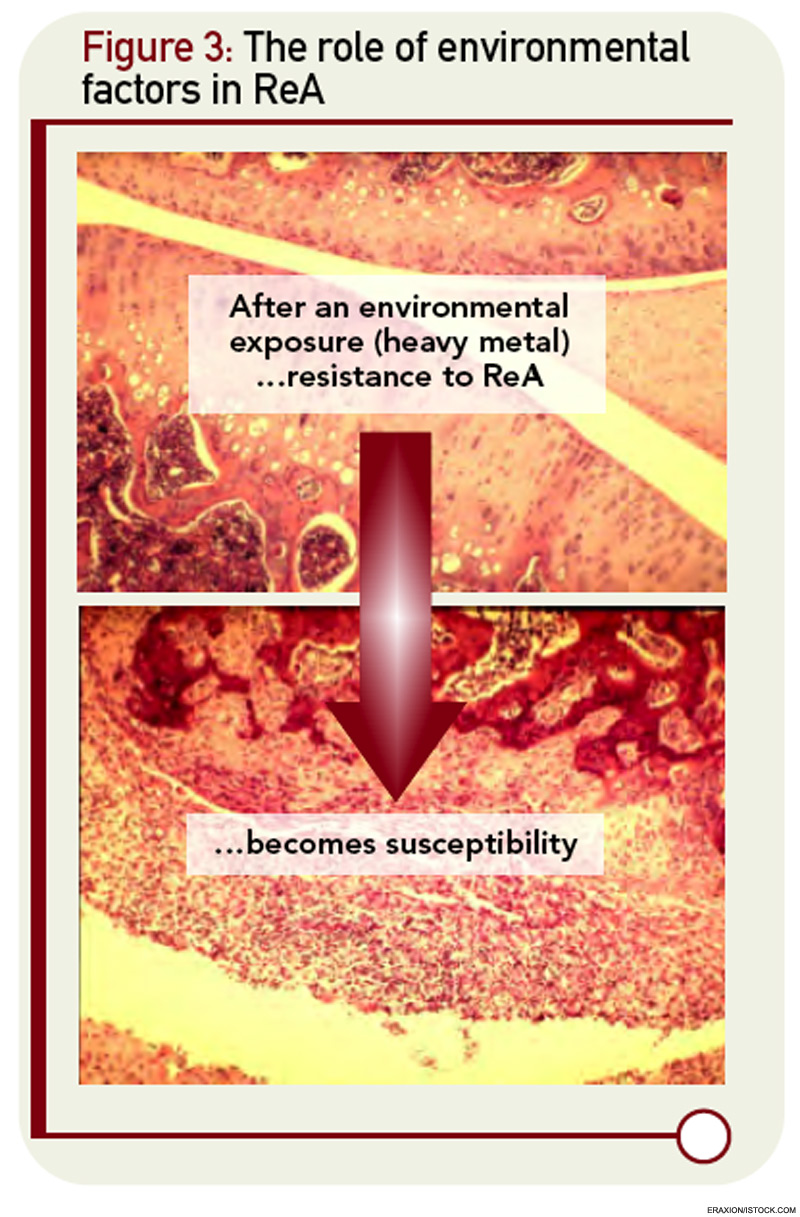
The underlying assumption in the molecular mimicry theories, exemplified by the studies of rheumatic fever, is that the host immune response to infection is overactive in nonseptic sequelae like ReA. But there are intriguing clues that host defenses might be diminished rather than amplified in patients with ReA. The cytokine profiles of ReA patients suggest down-regulation of proinflammatory cytokines rather than upregulation.17 These changes include a decrease in levels of TNF-α and IFN-γ, cytokines normally thought to be deleterious rather than beneficial cytokines with respect to arthritis. The role of cytokine dysregulation in disease etiology is borne out in experimental ReA.18 Animals that are susceptible to Chlamydia-induced arthritis have diminished rather than enhanced production of TNF-α and IFN-γ within the joint. This appears to relate to impaired capacity of host clearance of the organism in susceptible strains of animals. Of interest, this genetically defined cytokine signature can be dramatically altered by exposure to heavy metals, suggesting that other environmental factors may come into play in the dynamics of ReA in the clinical setting (see Figure 3, below right).19
Recent studies in CD have supported the role of defective host immune response to foreign organisms in immunopathogenesis. Genetic susceptibility to CD has been related to polymorphisms in the CARD15 gene, but the implicated SNPs suggest a loss-of-function process that would predict diminished, not enhanced, elaboration of TNF-α. This situation seems counterintuitive in a disease that is marked by TNF-mediated inflammation and that is responsive to anti-TNF agents. Recent studies have shed light on this paradox.20 Macrophage function in CD is indeed diminished, with impaired generation of proinflammatory cytokines, as a function of lysosomal degradation of precursor molecules, which impairs their egress to the secretory pathways of the macrophage. The net effect of this cytokine signature is a diminished inflammatory response to pathogens, with persistence of the organism being the consequence. Thus, the model for CD, as that for ReA, is undergoing a conceptual change, suggesting that therapeutic enhancement rather than diminution of host immune responses as a strategy for intervention.
Have we thus come full circle? Metchnikoff had postulated that the macrophage is the critical gatekeeper in host–pathogen interactions. With the expanded knowledge of innate immunity in arthritis and autoimmunity, and the recognition that TLR function as gatekeepers of adaptive immunity by upregulation of costimulatory molecules, the microbial aspects of ReA and CD have begun to be clarified. The genetic complexity of CD revealed in recent genome-wide association studies suggests that modulation of host–microbial interactions is a complex and changing story. But more pieces of this challenging jigsaw puzzle are now in place.
The Strange Case of Chlamydia-induced ReA
The recognition that Chlamydia trachomatis and Chlamydia pneumoniae species may exist in a persistent metabolically active infection state in the synovium suggests that persistent Chlamydiae may be susceptible to antimicrobial agents. One recent study was a nine-month, double-blind prospective trial assessing a six-month course of combination antibiotics as a treatment for Chlamydia-induced ReA.21 Treatment arms consisted of doxycycline and rifampin, azithromycin and rifampin, or placebo. After six months of treatment, 17 out of 27 subjects (63%) on antibiotics were responders compared with three of 15 (20%) on placebo. Antibiotic treatment was associated with significant improvement in the modified swollen joint count and tender joint count. There were significantly more subjects who became polymerase chain reaction negative at month six in the active therapy group than in the placebo group. These data suggest that a six-month course of combination antibiotics may be an effective therapy for chronic Chlamydia-induced ReA.
This study has raised important issues in several contexts. First, there is the conceptual issue that intraarticular persistence of pathogens may trigger a chronic inflammatory process that may be sustained, possibly for years. Secondly, the ambiguity in nomenclature becomes readily apparent, as it has been the case for Lyme arthritis. If the inflammatory process is terminated with antibiotic treatment, it could be argued that the treated condition meets a reasonable operational definition of septic arthritis, rather than reactive arthritis. Finally, the clinical implications are significant. The subjects in this study had a long-standing seronegative arthritis that had proved refractory to both nonsteroidal antiinflammatory drugs and disease-modifying anti-rheumatic drug treatment. This raises a very pertinent clinical question: How many patients currently classified as undifferentiated SpA might be effectively treated with antibiotics? It is a question for which Dr. Osler would have urged an answer.
Self/Nonself Complexities Arising
In the original formulation of the underlying principles of the innate immune response, perfect self/nonself discrimination was proposed as the one of the cardinal features because the pathogen associated molecular patterns (PAMPs) recognized by host innate immune receptors were so different from endogenous host ligands so as to never allow a case of mistaken identity. A PAMP was the molecular passport of an alien.
But what should happen in the event that an alien has not only invaded our home and native land, but has become enculturated and fully assimilated into the society? Such was the case with the bacterial symbionts of antiquity that invaded eukayotic cells, set up distinct societies, and in time became mitochondria. But, a genetic footprint of this event was left behind: our mitochondrial DNA retains significant sequences of bacterial DNA.22 With respect to immune recognition, this is far more profound—and more unnerving—than molecular mimicry of host and microbial determinants. This is molecular identity of host and microbial determinants.
What would transpire if the mitochondrial DNA were released systemically, as might occur following trauma? That might occasion a response that was indistinguishable from sepsis, which seems unlikely—yet that is precisely what occurs. The binding of mitochondrial DNA to TLR9 on the surface of neutrophils initiates activation of neutrophils and release of IL-8 in a way that looks just like an infection (see Figure 2, above left).23 Indeed, infusion of mitochondrial DNA alone initiates an intense pneumonitis similar to that seen in the systemic inflammatory response syndrome that occurs after trauma. This recognition is changing the landscape of the concept of infection. Perhaps the enemy has been lurking within all along. With respect to host immune response, perhaps we have met the enemy—and it is us.
As our thinking of self/nonself proved in retrospect to be simplistic, and the gap between infection and trauma begins to narrow, potential new scenarios arise in the pathogenesis of inflammatory joint disease. Could exacerbation of AS after trauma reflect such processes? Could chronic inflammatory events in the skin, with local tissue injury, set the stage for psoriatic arthritis? Could the mucosal injury of Salmonella gastroenteritis or Chlamydia urethritis invoke these pathways? Could all arthritis be reactive? These are fascinating questions. As the landscape is changing, we should stay tuned.
Dr. Inman is senior scientist in the Division of Genetics and Development at Toronto Western Research Institute and professor of medicine and immunology at the University of Toronto in Ontario, Canada.
References
- Zabriskie JB, Hsu KC, Seegal BC. Heart-reactive antibody associated with rheumatic fever: Characterization and diagnostic significance. Clin Exp Immunol. 1970; 7:147-159.
- van de Rijn I, Zabriskie JB, McCarty M. Group A streptococcal antigens cross-reactive with myocardium. Purification of heart-reactive antibody and isolation and characterization of the streptococcal antigen. J Exp Med. 1977;146:579-599.
- Khanna AK, Nomura Y, Fischetti VA, Zabriskie JB. Antibodies in the sera of acute rheumatic fever patients bind to human cardiac tropomyosin. J Autoimmun. 1997;10:99-106.
- Albert LJ, Inman RD. Molecular mimicry and autoimmunity. New Eng J Med. 1999;341:2068-2074.
- Lundberg K, Kinloch A, Fisher BA, et al. Antibodies to citrullinated alpha-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum. 2008;58:3009-3019.
- Schwimmbeck PL, Yu DT, Oldstone MB. Autoantibodies to HLA B27 in the sera of HLA B27 patients with ankylosing spondylitis and Reiter’s syndrome. Molecular mimicry with Klebsiella pneumoniae as potential mechanism of autoimmune disease. J Exp Med. 1987;166:173-181.
- Hermann E, Yu DT, Meyer zum Büschenfelde KH, Fleischer B. HLA-B27-restricted CD8 T cells derived from synovial fluids of patients with reactive arthritis and ankylosing spondylitis. Lancet. 1993;342:646-650.
- Popov I, Dela Cruz CS, Barber BH, Chiu B, Inman RD. Autoreactive anti-B27 CTL induced by in vitro stimulation with Chlamydia. J Immunol. 2002;169:4033-4038.
- Granfors K, Jalkanen S, von Essen R, et al. Yersinia antigens in synovial-fluid cells from patients with reactive arthritis. N Engl J Med. 1989;320:216-221
- Zhang X, Pacheco-Tena C, Inman RD. Microbe hunting in the joints. Arthritis Rheum. 2003;49:479-482.
- Tsui FWL, Xi N, Rohekar S, et al. TLR-2 variants are associated with acute reactive arthritis. Arthritis Rheum. 2008;58:3436-3438.
- Hammer RE, Maika SD, Richardson JA, Tang JP, Taurog JD. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: An animal model of HLA-B27-associated human disorders. Cell. 1990;63:1099-1112.
- Taurog JD, Richardson JA, Croft JT, et al. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med. 1994;180:2359-2364.
- Abdollahi-Roodsaz S, Joosten LA, Koenders MI, et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest. 2008;118:205-216
- Rahman P, Inman RD, Gladman D, Reeve J, Peddle L, Maksymowych W. Association of IL-23R variants and ankylosing spondylitis. Arthritis Rheum. 2008;58:1020-1025.
- Ciccia F, Bombardieri M, Principato A, et al. Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheum. 2009;60:955-965.
- Bas S, Kvien TK, Buchs N, Fulpius T, Gabay C. Lower level of synovial fluid interferon-gamma in HLA-B27-positive than in HLA-B27-negative patients with Chlamydia trachomatis reactive arthritis. Rheumatology (Oxford). 2003;42:461-467.
- Inman RD, Chiu B. Cytokine profiles in the joint define pathogen clearance and severity in Chlamydia-induced arthritis. Arthritis Rheum. 2006;54:499-507.
- Inman RD, Chiu B. Heavy metal exposure reverses genetic resistance to Chlamydia-induced arthritis. Arthritis Res Ther. 2009;11:19.
- Smith AM, Rahman FZ, Hayee B, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med. 2009;206:1883-1897.
- Carter JD, Espinoza LR, Inman RD, et al. Combination antibiotics as a treatment for chronic Chlamydia-induced reactive arthritis. Arthritis Rheum. 2010;62:1298-1307.
- Gray MW, Burger G, Lang BF. The origin and early evolution of mitochondria. Genome Biol. 2001;2:1018.
- Zhang Q, Raoof M, Chen Y, Sumi et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104-1047.