Right: The small molecule structure of aspirin (21 molecules). Left: A monoclonal antibody made up of 10,000–20,000 molecules.
DNA Illustrations / Science Source
As questions about biosimilar medications swirl among U.S. rheumatologists, a recently released report sheds some light on the European experience with biosimilars—and may offer some important insights for the U.S. market.
The report, Biosimilars in the EU: Information Guide for Healthcare Professionals, was released in late April by the European Medicines Agency (EMA) and the European Commission.1 Highlights:
- Biosimilars have been in the European Union (EU) since 2006, and the EU has approved the highest number of biosimilars worldwide.
- The EU monitoring system for safety concerns has not identified any relevant difference in the nature, severity or frequency of adverse effects between biosimilars and their reference medicines since their approval.
- Although biosimilars may have minor differences from their reference medicines, these differences are not clinically meaningful. There are no expected differences in safety and efficacy.
- Biosimilars in the EU have been approved using the same standards of pharmaceutical quality, safety and efficacy as those of all biological medicines.
- A biosimilar can rely on the safety and efficacy gained during research with the reference medicine. This means clinical trials are not needed for each indication for which the biosimilar seeks approval.
- Because an approved biosimilar is very similar to its reference medicine and has the expected comparable safety and efficacy in one therapeutic indication, data can be extrapolated to other indications for which the reference medicine has been approved. “Extrapolation needs to be supported by all the scientific evidence generated in comparability studies,” according to the report.
- Biosimilar competition offers benefits to EU healthcare systems because it can improve patient access to safe and effective medicines with proved quality.
The findings from the report demonstrate Europe’s 10-year head start compared with the U.S. market, says Angus B. Worthing, MD, FACP, FACR, Arthritis & Rheumatism Associates, in the Washington, D.C., area, and chair of the ACR’s Government Affairs Committee. “Biosimilars were first approved in Europe more than 10 years ago and with more than 25 drugs in use, none has been pulled for safety problems. Pharmacovigilance programs have not identified any safety issues in biosimilars that differed from reference drugs,” he says.
This broad experience contrasts with that in the U.S., which has just six biosimilars approved by the U.S. Food and Drug Administration (FDA) (see Table 1).
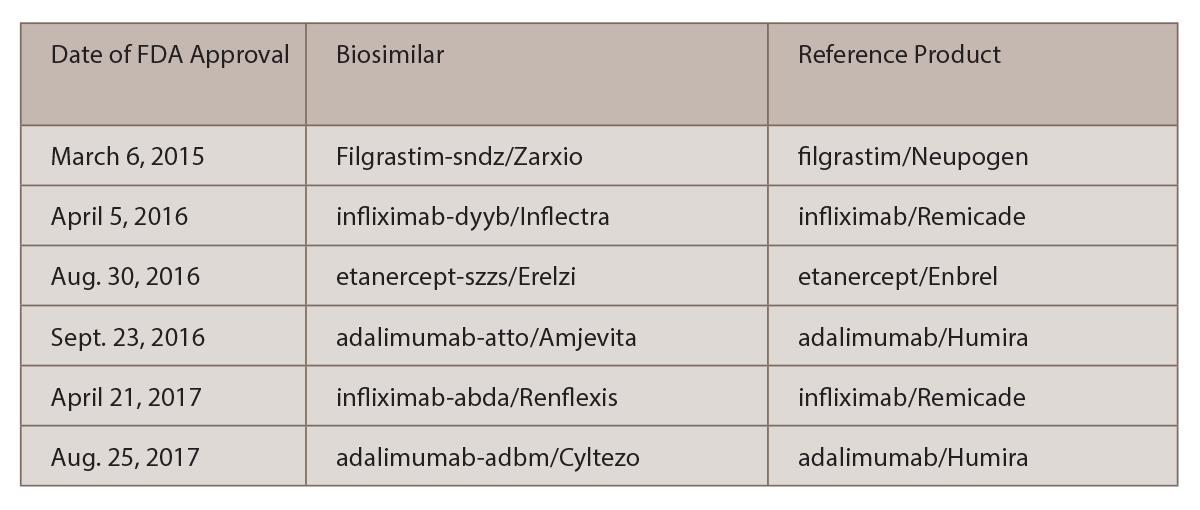
(click for larger image)
TABLE 1: U.S.-Approved Biosimilars
“[The EU has] had over 10 years of monitoring for any difference between biosimilars and originators, and they haven’t found anything to raise a red flag. They’ve been at the forefront,” says Stanley Cohen, MD, clinical professor, Department of Internal Medicine, University of Texas Southwestern Medical Center and in private practice, Rheumatology Associates, Dallas. “In most of the countries, the health departments are promoting the use of biosimilars as initiating therapy and haven’t determined any issues in terms of switching therapies.”
Safety & Efficacy
Both the report and the overall experience among European physicians indicate no specific safety or efficacy concerns for biosimilars compared with their reference products, says Jonathan Kay, MD, professor of medicine, Timothy S. and Elaine L. Peterson Chair in Rheumatology, and director of clinical research, Division of Rheumatology, University of Massachusetts Medical School, Worcester.
“If a biosimilar is truly biosimilar—and the FDA has strict requirements to demonstrate that—it should have the same safety profile as the parent compound,” says Ellen Gravallese, MD, chief, Division of Rheumatology, professor of Medicine, and Myles J. McDonough Chair in Rheumatology, University of Massachusetts Medical School. Dr. Gravallese is also the ACR secretary.
Efficacy is another aspect on which biosimilars and their reference products seem to match closely. “Comparative effectiveness studies have shown that biosimilars are equivalent to their reference products. When I have spoken with rheumatologists from around the world, no one has mentioned lesser efficacy of a biosimilar,” Dr. Kay says.
The only mentions of lesser efficacy of a biosimilar that he has seen were in several posters from Turkey that were presented at the 2016 ACR/ARHP Annual Meeting and at the 2017 European League Against Rheumatism (EULAR) Annual Meeting, which suggested that Celltrion’s biosimilar, infliximab-dyyb (Inflectra), was less effective than Remicade. Of note, the analyses reported in these posters were supported by the maker of Remicade.
“Despite the positive experience regarding safety and efficacy of biosimilars elsewhere, it is not unexpected that rheumatologists in the U.S. may have questions about biosimilars,” says Dr. Kay. “Because they haven’t had a lot of experience with biosimilars, they remain uncertain about their safety and efficacy,” he says.
There is confusion among physicians surrounding biosimilars and biomimics, and that can stir up more questions. Although biomimics are replicas of the reference product, they have not gone through the extensive regulatory review process to which biosimilars have been subjected, Dr. Kay adds. “Once a biosimilar has been reviewed and approved by regulators in a highly regulated area, one can be confident that it meets standards of efficacy and equivalence to its reference product,” he says. In contrast, some biomimics do not even have the same amino acid sequence as the reference products they intend to copy.
Interchangeability
Two major areas of interest with regard to biosimilars both in Europe and the U.S. are interchangeability and extrapolation of indications. With interchangeability, when a biologic is prescribed, the patient may actually end up getting a biosimilar. The question is, who can or should make that decision on interchangeability?
“A key difference between the EU and the U.S. biosimilars markets is how they address interchangeability,” Dr. Worthing says. “The EMA does not decide whether a drug is interchangeable with its reference product but rather leaves that decision—and thus, whether a pharmacy can substitute biosimilars automatically—to specific member nations. In the U.S., the FDA has proposed a separate approval process for interchangeable biosimilars that will require switching studies.”
“Biosimilars are not ‘knock-offs’ of their reference products. They are reverse engineered, designed and produced to be as close to the reference product as might be another batch of the reference product,” Dr. Kay says. The evidence so far seems to indicate that interchangeability would be reasonable because biosimilars are so similar to their reference product.
“A big question is how safe it will be to switch from the originator to a biosimilar,” Dr. Cohen says. “You will have nonmedical switching, in which the insurance company will say [a biologic] is not on their formulary or it’s a tier 3 drug, so you must use a biosimilar. I don’t think that will be an issue. We have multiple data that allow for a single switch, and there’s no issue.”
The ACR “strongly supports the FDA’s proposal to require manufacturers use robust switching studies to determine whether alternating between a biosimilar and its reference product impacts the safety or efficacy of the drug. Such studies are vital to achieve a clear understanding of what patients are likely to experience with changing formularies in a multi-payer, multi-state, and ever-changing market. These data should be open and accessible for outside investigators to analyze, and should be made clearly available to prescribing doctors in the interchangeable drug’s label via text or hyperlink. Further, we urge the FDA to consider requiring manufacturers to submit updated and standardized pharmacovigilance data as a prerequisite to certain post-market labeling changes.”
Dr. Cohen heard a colleague in Europe speculate whether psychologically, patients feel that a drug that is cheaper to use is not as effective as the more expensive counterpart. It’s also common for biologics to undergo very minor manufacturing changes as each batch is produced; although that has not been shown to change the effectiveness of the medication.
Another area of concern is extrapolation of indications, which the European report notes is an approach that has been used over the past decade.1 “If a biosimilar is highly similar to its reference product and has comparable safety and efficacy in one therapeutic indication, safety and efficacy data may be extrapolated to other indications for which the reference medicine is approved,” according to the report. By adhering to this concept, fewer clinical trials are needed to allow use of the biosimilar to be expanded to other disease states.
“It doesn’t make sense to require multiple trials [for extrapolation],” Dr. Cohen says. “Most of the work has been done with assays that determine this is a highly similar molecule, with no impact on efficacy or safety.” Although this may involve some griping from certain specialists—say, gastroenterologists who may not have data available from specific studies of the biosimilar conducted in patients with ulcerative colitis or Crohn’s disease—Dr. Cohen points out that trials within rheumatoid arthritis (RA) and psoriasis have established outcome measures that are considered more sensitive to detect a difference in the biosimilar and the reference molecule than inflammatory bowel disease, which is why sponsors have chosen to test their biosimilars in RA and psoriasis.
The ACR states that it “does not support automatic extrapolation. We do believe that extrapolation should be rigorously studied and fully utilized to help reduce the cost of these drugs. Throughout the biosimilar approval process, care must be taken to ensure that a drug pursuing interchangeability has successfully demonstrated extrapolation for all indications for which the originator is approved.”
Extrapolation of indications is supported by 10 years of safety and efficacy experience in Europe, Dr. Worthing says.
Pricing
Naturally, pricing of biosimilars is an issue of concern for insurers, manufacturers, physicians and patients. Dr. Gravallese believes that pricing is an important topic to address in any U.S. analysis of the role of biosimilars, including the upcoming ACR white paper.
“I would like to see biosimilars be used, because this will potentially lead to a lower medication cost. That’s something many of us would like to see happen,” Dr. Gravallese says.
One advantage of biosimilars so far is their cost, typically 15% lower than their reference product; however, Dr. Kay has seen examples of reference products that have been discounted to below the price of their biosimilars. “If that happened all the time, it might drive biosimilars out of the market. The survival of biosimilars is necessary to maintain a competitive marketplace and keep down the cost of effective therapies so that they are affordable for our patients,” he says.
In June, the U.S. Supreme Court overturned a lower court’s ruling that prevented Novartis from selling the biosimilar Neuopogen for six months after the U.S. FDA approval. That ruling is expected to help acceleration the entry of more biosimilars into the U.S. market, many predict.
ACR White Paper
To help address the issues mentioned above in the U.S. market, the ACR decided to produce a white paper on the topic of biosimilars. The rheumatologists quoted in this article are involved in the production of the white paper, as are a handful of others. Although there was originally talk about writing a review article on biosimilars, a paper that focuses on concerns specific to the use of biosimilars in the U.S. would likely be more beneficial, Dr. Gravallese points out.
“The ACR’s white paper on biosimilars will inform prescribers about the FDA approval process, scientific and clinical issues, such as extrapolation, immunogenicity, switching and interchangeability; and issues related to cost, access and the mechanics of prescribing,” Dr. Worthing says. “The purpose of the white paper is to inform our members about biosimilars, reassure them about the scientific rigor used by regulatory authorities and boost confidence in prescribing biosimilars.”
As of publication, this paper was still in draft form.
Vanessa Caceres is a medical writer in Bradenton, Fla.
Reference
- European Medicine Agencies and European Commission. Biosimilars in the EU: Information guide for healthcare professionals. 2017.