Image Credit: Stepan Kapl/shutterstock.com
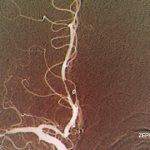
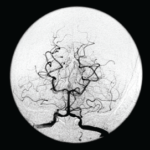
SAN FRANCISCO—Let’s say your radiologist comes to you and says that an angiogram gives a diagnosis of CNS vasculitis on four patients, all with acute onset of headache and stroke: One is a 25-year-old woman who is three months pregnant. Another is a 50-year-old man using excessive doses of nasal decongestants. Another is a 40-year-old woman with uncontrolled migraines. And then there’s a 30-year-old man who snorts cocaine.
There’s a problem here, said Julius Birnbaum, MD, MHS, assistant professor of neurology at Johns Hopkins, who is board certified in both rheumatology and neurology: “These patients do not have clinical features which are typical of CNS [central nervous system] angiitis.” Instead, he said, they likely have reversible vasoconstriction syndrome (RVCS), in which the underlying pathophysiology is a vasospasm, not vasculitis.
The unreliability of angiogram diagnoses was one of the points Dr. Birnbaum emphasized in his talk on rheumatic diseases that are manifest in the central nervous system, part of a clinical review course held at the 2015 ACR/ARHP Annual Meeting. He discussed primary angiitis of the CNS (PACNS), CNS syndromes in neuropsychiatric systemic lupus erythematosus (NPSLE) and demyelinating syndromes in Sjögren’s syndrome (SS).
PACNS

Dr. Birnbaum
Angiograms, Dr. Birnbaum said, are “notoriously non-specific,” and the same signs “can be seen in a plethora of non-inflammatory vasculopathies.”