Aksabir/shutterstock.com
Rheumatoid arthritis (RA) is rarely associated with renal manifestations, but secondary amyloidosis due to chronic inflammation is reported to be the etiology of renal dysfunction in many cases of RA.1,2
The discovery of biologic therapy, with TNF-alpha inhibitors in particular, made a huge difference in the disease course and prognosis of RA patients. However, TNF-alpha inhibitor use is linked with paradoxical development of systemic or organ-specific autoimmune processes (e.g., lupus, vasculitis, interstitial lung disease, multiple sclerosis, autoimmune hepatitis, inflammatory myopathies, peripheral neuropathies).3,4
Case Presentation
A 69-year-old African American female with 25 years’ history of seropositive, erosive RA presented to the hospital with complaints of abdominal discomfort, bloating, constipation and loss of appetite. In the year prior to this admission, pain and swelling in her hands, wrists, elbows and ankles were accompanied by two hours of morning stiffness. Physical examination revealed an ill-appearing woman in moderate discomfort, without rashes, but with bilateral metacarpophalangeal (MCP) and proximal interphalangeal (PIP) synovitis, swan neck deformities, bilateral ulnar deviation and mild knee effusions and hammertoes. In addition, rheumatoid nodules over her second and third right PIPs and left elbow were noted. Two-plus pedal edema was present up to knee level.
Renal dysfunction pathophysiology in RA patients is frequently linked to secondary amyloidosis due to long-lasting chronic inflammation. Rarely, the etiology of renal involvement is reported to be secondary to renal vasculitis or an autoimmune process.
Review of her medical chart revealed that, over the years, the patient had failed multiple disease-modifying anti-rheumatic drugs (DMARDs), such as hydroxychloroquine, methotrexate and leflunomide, and several biologic medications, including infliximab, etanercept and adalimumab. The last time she had received biologic treatment was approximately one year prior to this admission.
At the time of hospitalization, the patient was treated with 10 mg of prednisone daily. For hypertension, the patient was receiving 100 mg hydralazine three times per day.
A laboratory workup revealed that the patient was anemic (Hb 9.7 g/dL; normal range 12–14 g/dL) and leukopenic (WBC 3.7 10*3/uL; normal range 3.9–10.8 10*3/uL), and markers of inflammation (CRP 20 mg/dL; normal range <10 mg/dL; ESR 71 mm/hr; normal range <20 mm/hr) were significantly elevated.
In the previous year, her creatinine increased steadily from 1.2 to 2.3 mg/dL (normal range 0.50–1.20 mg/dL) at the time of admission. Additionally, nephrotic range proteinuria was noted (urine protein/creatinine ratio = 3,230 mg/g). Immunology workup was positive for ANA (homogeneous pattern, titer of 1:320), anti-histone antibodies (moderate 1.6; normal range <1 negative), CCP (>250 units, normal range <20 units), rheumatoid factor (506 IU/mL; normal range <14 IU/mL) and SSA/SSB >8 antibodies (normal range; negative).
In contrast, cryoglobulins, dsDNA, Sm/RNP, ANCA (PR-3, MPO), anticardiolipin, anti-B2-glycoprotein antibodies and lupus anticoagulant screens were negative. The infectious workup included HIV and hepatitis A, B and C tests that were also negative.
An anemia workup was suggestive of anemia of chronic disease. Further testing included serum electrophoresis that did not reveal any monoclonal spike. Interestingly, the subtypes of IgG found a mildly decreased serum IgG1 (333 mg/dL; normal range 382–929 mg/dL). Both C3 and C4 serum complements were significantly reduced (C3 57 mg/dL; normal range 83–193 mg/dL; C4 -4 mg/dL; normal range 15–57 mg/dL).
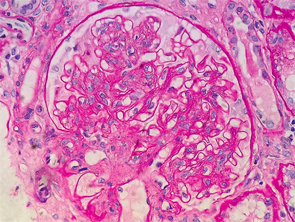
Figure 1: Renal biopsy. H&E staining: enlarged glomeruli, mesangial expansion with diffuse increased cellularity; glomerular basement membranes irregularly thickened, but no vasculitis, necrotizing lesions, crescents or hyaline thrombi were seen within the capillary loops.
Creatinine worsening accompanied by nephrotic range proteinuria, low serum complements levels, positive ANA, SSA/SSB and anti-histone antibodies in the context of active RA disease suggested an autoimmune process as a plausible cause of current renal failure.
Initial fluid resuscitation resulted in no improvement of the patient’s renal function. The decision to pursue renal biopsy was made after consultation with the nephrology service.
Interestingly, the renal biopsy was consistent with immune complex-mediated MPGN. Pathology reported enlarged glomeruli, mesangial expansion with diffuse increased cellularity; the glomerular basement membranes were irregularly thickened, but no vasculitis, necrotizing lesions, crescents or hyaline thrombi were seen within the capillary loops (see Figure 1).
Immunofluorescence staining revealed predominant IgG and C3 deposition in the glomerular basement membrane and mesangial compartment. Additionally, with lower predominance the immunostaining was positive for IgM and IgA deposition. However, immunostaining was negative for C1q or hyaline thrombi usually seen in lupus nephritis and renal vasculitis, respectively. Electron microscopy confirmed the irregular thickening of the glomerular basement membrane, with a few scattered subendothelial electron dense deposits, focal neomembrane formation, an expanded mesangium and podocyte foot processes effacement (see Figure 2).
The patient was treated with high-dose pulse steroids for three days (methylprednisolone 1 g/day), followed by prednisone 1 mg/kg/day. Hydralazine was discontinued shortly after admission and replaced with amlodipine. While in the hospital, she received the first dose of Rituximab (1,000 mg), followed by a second dose 14 days later. On Day 7 of admission, the patient’s complaints subsided, and her creatinine was noted to decrease to 1.55 mg/dL.
The patient was discharged on 60 mg/day of prednisone with the recommendation to be slowly tapered as an outpatient. Table 1 (below) depicts the values of creatinine and proteinuria at admission, discharge and two-, six- and nine-month follow-up points. Over time, both renal function and level of proteinuria improved significantly.
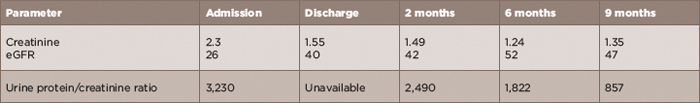
TABLE 1: Creatinine (mg/dL), eGFR (mL/min/1.73 m2) and urine protein/creatinine ratio (mg/g) at admission, discharge,
2-, 6- and 9-month follow-up
Discussion
Renal dysfunction pathophysiology in RA patients is frequently linked to secondary amyloidosis due to long-lasting chronic inflammation.1,2 Rarely, the etiology of renal involvement is reported to be secondary to renal vasculitis or an autoimmune process. A review of literature resulted in sparse case reports of RA patients who developed minimal change disease, mesangial proliferative, focal segmental/proliferative or fibrillary glomerulonephritis (GN).6
Drug-induced glomerulopathies are most likely immune mediated, and they can present with a nephritic or nephrotic syndrome. Focal segmental glomerulosclerosis, mesangial crescentic and MPGN have been reported. Patients with HLA-B8 and DR3 antigens are more susceptible for drug-induced GN.7,8
In an effort to find if she is predisposed to develop a drug-induced autoimmune GN, our patient was HLA typed. She carried the following HLA class I antigens: A1, B8, B62, C3, C7 and class II antigens DR4, DR17, DQA1 03; 05, DQB1 02; 03. As previously mentioned, carriers of HLA-B8 antigens are predisposed to drug-induced GN, and HLA-DR4 antigens are known to be slow acetylators.9
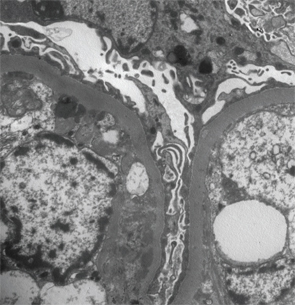
Figure 2: Renal biopsy. Electron microscopy: irregular thickening of the glomerular basement membrane with a few scattered subendothelial electron dense deposits, focal neomembrane formation, an expanded mesangium and podocyte foot processes effacement.
Exposure to TNF-alpha inhibitors, particularly in RA, is associated with membranous or necrotizing crescentic GN.6 Piga et al performed a review of the literature of patients with chronic inflammatory rheumatic diseases treated with biologics who developed autoimmune renal disorders.10 Interestingly, 22 out of 29 cases were RA patients, with Etanercept being the cause in about 51% of cases, followed by adalimumab (31.0%), infliximab (10.3%), tocilizumab and abatacept (3.4% each).10 Drug-induced lupus due to TNF-alpha inhibitors manifested clinically with rash (73%), polyarthritis (52%), fever (52%), myalgias (24%), pericardial effusion (9%) and, rarely, with nephritis (9%).3,4
Common features in TNF-alpha drug-induced GN were proteinuria, hypocomplementemia and positive SSA antibodies, although in the majority of cases, the renal dysfunction was reported during the time of the therapy with significant improvement after discontinuation.4,11
Approximately one year from last exposure to TNF-alpha inhibitors, our patient presented with severe arthritis, nonspecific complaints and renal insufficiency accompanied by nephrotic range proteinuria without hematuria. Serologically, she was positive for ANA, anti-histone and SSA/SSB antibodies, but negative for dsDNA or cryoglobulinemia.
In this scenario, hypocomplementemia was suggestive of accelerated consumption by immune complex formation. The renal biopsy confirmed our suspicion of ongoing autoimmune process, demonstrating immune-complex deposition MPGN.
The challenge of this case was the patient’s previous exposure to hydralazine, a drug known to induce lupus; however, our patient did not present any of the classic signs of lupus, and her renal biopsy was not consistent with lupus nephritis; staining for C1q deposition was negative.8
Drug-induced glomerulopathies are most likely immune mediated, & they can present with a nephritic or nephrotic syndrome.
Rituximab, a monoclonal antibody to the CD20 antigen on B cells, was approved in 2007 for RA patients who failed DMARDs and TNF-alpha inhibitors. Additionally, in 2011, it was approved for ANCA vasculitis (RAVE trial), because it demonstrated non-inferiority to cyclophosphamide in patients with renal involvement.12,13 It is also used off-label in many immune-mediated GN cases, including lupus nephritis, membranous nephropathy, mixed cryoglobulinemia, minimal disease or focal segmental glomerulosclerosis given its efficacy, tolerability and safety profile.14
Our patient was started on Rituximab, with significant improvement in her arthritis symptoms and renal function.
Conclusion
This is a case of a long-term, poorly controlled RA, with exposure to multiple TNF-inhibitors that developed late-onset immune complex-mediated membrano-proliferative glomerulonephritis. Treatment with high-dose pulse steroids and Rituximab resulted in significant improvement of the patient’s renal function and reduction of proteinuria.
 Diana M. Girnita, MD, PhD, is a rheumatology fellow at the University of Cincinnati in the Department of Immunology, Allergy and Rheumatology.
Diana M. Girnita, MD, PhD, is a rheumatology fellow at the University of Cincinnati in the Department of Immunology, Allergy and Rheumatology.
Shahzad Safdar, MD, is a nephrologist at The Christ Hospital in Cincinnati. He specializes in electrolyte abnormalities, hypertension, kidney stones, chronic kidney disease, dialysis, glomerulonephritis including lupus nephritis and other connective tissue-related renal diseases. He has been an attending nephrologist at The Christ Hospital since 1998.
Avis Ware, MD, is a professor of medicine at the University of Cincinnati in the Department of Immunology, Allergy and Rheumatology. She is board certified in both internal medicine and rheumatology. Her research interests lie in general rheumatology and integrative medicine.
References
- Helin HJ, Korpela MM, Mustonen JT, Pasternack AI. Renal biopsy findings and clinicopathologic correlations in rheumatoid arthritis. Arthritis Rheum. 1995 Feb;38(2):242–247.
- Pruzanski W. Renal amyloidosis in rheumatoid arthritis. J Rheumatol. 2007 Apr;34(4):889; author reply 889.
- Perez-Alvarez R, Pérez-de-Lis M, Ramos-Casals M, BIOGEAS study group. Biologics-induced autoimmune diseases. Curr Opin Rheumatol. 2013 Jan;25(1):56–64.
- Costa MF, Said NR, Zimmermann B. Drug-induced lupus due to anti-tumor necrosis factor alpha agents. Semin Arthritis Rheum. 2008 Jun;37(6):381–387.
- Stokes MB, Foster K, Markowitz GS, et al. Development of glomerulonephritis during anti-TNF-alpha therapy for rheumatoid arthritis. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc—Eur Ren Assoc. 2005 Jul;20(7):1400–1406.
- Kronbichler A, Mayer G. Renal involvement in autoimmune connective tissue diseases. BMC Med. 2013 Apr 4;11:95.
- Izzedine H, Launay-Vacher V, Bourry E, et al. Drug-induced glomerulopathies. Expert Opin Drug Saf. 2006 Jan;5(1):95–106.
- Xiao X, Chang C. Diagnosis and classification of drug-induced autoimmunity (DIA). J Autoimmun. 2014 Feb–Mar;48–49:66–72.
- Clancy RM, Backer CB, Yin X, et al. Genetic association of cutaneous neonatal lupus with HLA class II and tumor necrosis factor alpha: Implications for pathogenesis. Arthritis Rheum. 2004 Aug;50(8):2598–2603.
- Piga M, Chessa E, Ibba V, et al. Biologics-induced autoimmune renal disorders in chronic inflammatory rheumatic diseases: Systematic literature review and analysis of a monocentric cohort. Autoimmun Rev. 2014 Aug;13(8):873–879.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore). 2007 Jul;86(4):242–251.
- Geetha D, Specks U, Stone JH, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis with renal involvement. J Am Soc Nephrol JASN. 2015 Apr;26(4):976–985.
- Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010 Jul 15;363(3):221–232.
- Kattah AG, Fervenza FC, Roccatello D. Rituximab-based novel strategies for the treatment of immune-mediated glomerular diseases. Autoimmun Rev. 2013 Jun;12(8):854–859.

