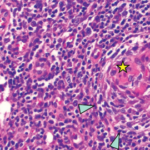
Seven months later, she was readmitted for cellulitis of the right arm and found to have a neck mass and a new systolic murmur, and was noted to have pitted scars on her fingertips; at that point, she fulfilled the 2013 ACR/EULAR classification criteria for systemic sclerosis. A CT of her neck revealed persistent lymphadenopathy, and a 2D echo showed persistently elevated pulmonary arterial pressure of 44 mmHg.
Discussion
We present the case of a patient with newly diagnosed SLE and MCD and who also has evidence of EBV reactivation and features of systemic sclerosis.
Castleman disease is a heterogeneous group of disorders with three histological types (i.e., hyaline vascular, plasma cell and mixed) and two clinical subtypes (i.e., localized and multicentric).1 Most cases of MCD occur in middle-aged men, and there is an association between MCD and HIV and human herpes virus 8.2,3 SLE, however, occurs mainly in women of childbearing age. The most common lymph node lesions in SLE are follicular hyperplasia and coagulative necrosis, with the latter being more specific.4
In one review of 33 SLE patients, lymph node pathology consistent with Castleman disease was found in 10% of these patients.4 The reported cases of concomitant MCD and SLE were mainly young to middle-aged women who were HIV negative, like our patient.5-10 Our patient was also noted to have evidence of EBV infection with IgM and IgG antibodies, as well as EBV DNA detected on PCR. These findings suggest that either her EBV infection was subacute or she was reinfected with EBV.11
EBV occurs worldwide, with over 80% of individuals older than 30 being infected. The infection persists for life and is associated with the development of malignancies, such as Burkitt’s lymphoma, Hodgkin’s disease and lymphoproliferative disorders.12 EBV has been reported in association with Castleman disease in variable frequencies, ranging from 2 out of 20 to 19 out of 20 cases.13,14 In one instance, EBV was reported to occur in MCD, with fatal results.15
IL-6 may be produced in an autocrine fashion from EBV-transformed B cells.16 Excessive IL-6 production in mice produces a syndrome characterized by anemia, transient granulocytosis, splenomegaly, hypoalbuminemia and polyclonal hypergammaglobulinemia, which closely resembles the systemic features of Castleman disease.17 The level of IL-6 produced by hyperplastic lymph nodes in patients with Castleman disease is directly related to the severity of clinical features and worsening of the lab results, which resolved with the removal of the lymph nodes.18