Jarun Ontakrai / shutterstock.com
Over the past few years, biosimilars and other new drugs have been introduced to treat rheumatic illnesses. Some of the conditions we treat have numerous drug options, others have few or only off-label options. This series, “Rheumatology Drugs at a Glance,” provides streamlined information on the administration of biologic, biosimilar and small molecule inhibitor drugs used to treat patients with rheumatic disease. In Part 3 of this series, we discuss the 21 available options to treat rheumatoid arthritis (RA; see Table 1, opposite).
RA
RA is a common chronic autoimmune disorder characterized by the simultaneous inflammation of the synovium of multiple joints that frequently leads to joint destruction, deformity, and disability.1,2 The estimated prevalence of RA increases with age in both sexes and is greatest in patients aged 40–70 years old. Productivity loss at work is associated with personal, clinical and work-related factors for patients with RA.3 At-work productivity loss is also negatively associated with health-related quality of life, especially in the areas of mental health, physical role limitations and pain.
The goals of treatment are to improve patients’ quality of life, reduce functional limitations, prevent joint damage and decrease disease complications. Treatment decisions should be made by the healthcare team and patients through a shared decision-making process, taking into account patients’ values and preferences, as well as their co-morbidities. It’s important to select the best therapeutic option for and with the patient to improve adherence and allow the patient to attain the highest level of satisfaction.
RA treatments can be expensive, and healthcare providers can help make treatment costs more manageable for patients. A number of financial assistance resources are available to patients, and patients can also contact the manufacturers to request co-pay assistance. Your staff can also explore patient assistance programs for those who do not have insurance (https://www.rheumatoidarthritis.org/resources).7
Depending on a patient’s healthcare coverage and the medication(s) they use, a specialty pharmacy may dispense the medications. Most specialty pharmacies have clinical pharmacists available who can help patients manage insurance issues, provide drug monitoring for adverse effects, effectiveness and adherence issues, and serve as a drug information resource.
For additional information, review the ACR’s published RA guideline.1 The ACR anticipates publishing an updated guideline in late 2019/early 2020.
It’s important to select the best therapeutic option for & with the patient to improve adherence & for the patient to attain the highest level of satisfaction.
The Drugs
 Abatacept (Orencia):8 injection/infusion
Abatacept (Orencia):8 injection/infusion
Drug class: selective T cell co-stimulation modulator, DMARD, immunomodulator
Warnings & Precautions
- Concomitant use with a tumor necrosis factor inhibitor (TNFi) can increase the risk of (serious) infections. Discontinue abatacept if a serious infection occurs.
- Anaphylaxis or anaphylactoid reactions can occur after the first infusion and can be life threatening. Appropriate medical support should be immediately available in the event of a reaction. Postmarketing experience reveals at least one case of fatal anaphylaxis following the first abatacept infusion. Following an anaphylactic or other serious allergic reaction, abatacept administration should be stopped immediately with appropriate therapy instituted. Abatacept should be permanently discontinued.
- Patients with a history of recurrent infections or underlying conditions predisposing them to infections may experience more infections.
- Screen for latent tuberculosis (TB) prior to initiating therapy. Patients testing positive should be treated prior to abatacept initiation.
- Live vaccines should not be given concurrently or within three months of abatacept discontinuation.
- Based on its mechanism of action, abatacept may blunt the effectiveness of some immunizations.
- Chronic obstructive pulmonary disease patients may develop more frequent respiratory adverse events.
Dosage & Administration
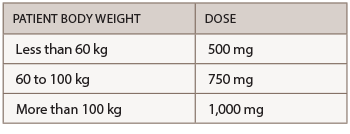
Abatacept is administered by subcutaneous (SQ) injection and intravenous (IV) infusion. The recommended frequency for SQ injection is once weekly, with or without an IV loading dose. For patients initiating therapy with an IV loading dose, administer a single IV infusion (per body weight categories provided in the table below), followed by the first 125 mg SQ injection given within a day of the IV infusion. Patients transitioning from IV therapy to SQ administration should receive the first SQ dose instead of the next scheduled IV dose.
The recommended dose for IV infusion is based on the patient’s body weight.
Commentary
The U.S. Food and Drug Administration (FDA) approved abatacept based on the results of two randomized, controlled trials that involved nearly 3,000 patients. The trials showed that abatacept inhibited joint damage progression independent of patients’ baseline clinical, biochemical or radiographic characteristics. The most common adverse reactions (≥10%) are headache, upper respiratory tract infection, nasopharyngitis and nausea.
Adalimumab (Humira):9 injection
Biosimilars: adalimumab-atto (Amjevita),10 adalimumab-adbm (Cyltezo),11 adalimumab-
adaz (Hyrimoz)12
Drug class: DMARD, TNFi
Boxed warning: Refer to important safety information (ISI) below, and
- Fatal hepatosplenic T cell lymphomas (HSTCLs) have occurred post-marketing in adolescent and young adult inflammatory bowel disease (IBD) patients treated with TNFi’s.
*Important Safety Information
This ISI is applicable to all tumor necrosis factor inhibitors (TNFi’s) and some other immune modulators.
Serious Infections & Malignancies
- There is an increased risk of serious infections leading to hospitalization or death, including TB, bacterial sepsis, invasive fungal infections (e.g., histoplasmosis) and infections due to other opportunistic pathogens. If these develop, discontinue the drug.
- Patients should be tested for latent TB prior to starting drug and during therapy. Treatment for latent TB should be initiated prior to starting the drug. Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with the drug, including the possible development of TB in patients who tested negative for latent TB infection prior to initiating therapy. Induration of 5 mm or greater with tuberculin skin testing should be considered a positive test result when assessing if treatment for latent TB is needed prior to initiating the drug, even for patients previously vaccinated with bacillus Calmette-Guerin (BCG).
- Lymphoma and other malignancies, some fatal, have been reported in children and adolescents treated with TNFi’s.
Warnings & Precautions
- Do not start adalimumab during an active infection. If an infection develops, monitor carefully and stop adalimumab if the infection becomes serious.
- Invasive fungal infections—for patients who develop a systemic illness on adalimumab, consider empiric anti-fungal therapy for those at high risk who reside in or travel to regions where mycoses, such as histoplasmosis, coccidioidomycosis or blastomycosis, are endemic.
- The risks and benefits of treatment with TNFi’s, including adalimumab, should be considered prior to initiating therapy in patients with a known malignancy, other than a successfully treated non-melanoma skin cancer, or when considering continuing a TNF blocker in patients who develop a malignancy.
- Anaphylaxis or serious allergic reactions may occur.
- Hepatitis B virus (HBV) reactivation can occur. Monitor HBV carriers during and several months after therapy. If reactivation occurs, stop adalimumab and begin anti-viral therapy.
- Demyelinating disease, exacerbation or new onset, may occur.
- Cytopenias, pancytopenia—advise patients to seek immediate medical attention if they develop signs and symptoms suggestive of blood dyscrasias or infection (e.g., persistent fever, bruising, bleeding, pallor) while on adalimumab. Consider discontinuation of therapy in patients with confirmed significant hematologic abnormalities.
- Heart failure, worsening or new onset, may occur.
- Lupus-like syndrome—stop adalimumab if the syndrome develops.
Dosage & Administration
Adalimumab is administered by SQ injection. The recommended dose is 40 mg every other week with or without methotrexate (MTX). Some RA patients not receiving MTX may benefit from increasing the frequency to 40 mg every week.
Commentary
The approval of adalimumab was based on four randomized, double-blind, clinical studies in more than 2,000 patients who had active RA. In all four studies, adalimumab showed significantly greater improvement than placebo on the disability index of the Health Assessment Questionnaire from baseline to the end of the study, and significantly greater improvement than placebo in health outcomes as assessed by the Short Form Health Survey. Improvement was also seen in both the physical and mental component summaries. The most common adverse reactions (≥10%) were infections (e.g., upper respiratory, sinusitis), injection-site reactions, headache and rash.
Certolizumab pegol (Cimzia):13 injection
Drug class: DMARD, TNFi
Boxed warning: Refer to *ISI (p. 14)
Warnings & Precautions
- Do not start certolizumab during an active infection. If an infection develops, monitor carefully and stop certolizumab if the infection becomes serious.
- Invasive fungal infections—for patients who develop a systemic illness on certolizumab, consider empiric anti-fungal therapy for those at high risk who reside in or travel to regions where mycoses, such as histoplasmosis, coccidioidomycosis or blastomycosis, are endemic.
- Cases of lymphoma and other malignancies have been observed in patients receiving TNFi’s.
- Heart failure, worsening or new onset, may occur.
- Anaphylaxis or serious allergic reactions may occur.
- HBV reactivation can occur; test for HBV infection before starting certolizumab. Monitor HBV carriers during and several months after therapy. If reactivation occurs, stop certolizumab and begin anti-viral therapy.
- Demyelinating disease, exacerbation or new onset, may occur.
- Cytopenias, pancytopenia—advise patients to seek immediate medical attention if symptoms develop, and consider stopping certolizumab.
- Lupus-like syndrome—stop certolizumab if the syndrome develops.
Strategies to achieve remission &/or low disease activity include initiating disease-modifying anti-rheumatic drug therapy early in the disease course to slow & prevent joint damage.
Dosage & Administration
Certolizumab is administered by SQ injection. The recommended initial dose is 400 mg (two SQ injections of 200 mg each) and 400 mg at Weeks 2 and 4, followed by 200 mg every other week. A maintenance dose of 400 mg every four weeks can be considered.
Commentary
The FDA approval was based on clinical trials that enrolled more than 2,300 patients. The results showed RA patients taking Cimzia with MTX had a greater reduction in signs and symptoms of RA after 24 weeks of treatment than patients taking only MTX. Some patients showed clinical responses to certolizumab plus MTX within one to two weeks. The most common adverse reactions (≥7%) were upper respiratory tract infection, rash and urinary tract infection.
Etanercept (Enbrel):14 injection
Biosimilars: etanercept-szzs (Erelzi),15 etanercept-ykro (Eticovo)16
Drug class: DMARD, TNFi
Boxed warning: Refer to *ISI (p. 14)
Warnings & Precautions
- Do not start etanercept during an active infection. If an infection develops, monitor carefully and stop etanercept if the infection becomes serious.
- Consider empiric anti-fungal therapy for patients at high risk (those who reside in or travel to regions where mycoses, such as histoplasmosis, coccidioidomycosis or blastomycosis, are endemic) of invasive fungal infections who develop a severe systemic illness on etanercept.
- Demyelinating disease, exacerbation or new onset, may occur.
- Cases of lymphoma have been observed in patients receiving TNFi’s.
- Congestive heart failure, worsening or new onset, may occur.
- Advise patients to seek immediate medical attention if symptoms of pancytopenia or aplastic anemia develop, and consider stopping etanercept.
- Monitor HBV carriers for reactivation during and following therapy. If reactivation occurs, consider stopping etanercept and beginning anti-viral therapy.
- Anaphylaxis or serious allergic reactions may occur.
- Stop etanercept if lupus-like syndrome or autoimmune hepatitis develops.
Dosage & Administration
Etanercept is administered by SQ injection. The recommended initial dose is 50 mg once weekly with or without MTX.
Commentary
Etanercept was the first TNFi approved for the treatment of RA. The results of the clinical trials have demonstrated its clinical safety and efficacy in early, as well as established, disease. Patients with RA on etanercept were found to not only have reduced disease activity, but also have limited progression of joint damage in early and late disease. Etanercept can be used as a monotherapy or in combination with MTX, although the combination therapy appears to be more effective in terms of reducing joint damage. The most common adverse reactions (≥5%) were infections and injection-site reactions.
Golimumab (Simponi):17 injection
Drug class: DMARD, TNFi
Boxed warning: Refer to *ISI (p. 14)
Warnings & Precautions
- Do not start golimumab during an active infection. If an infection develops, monitor carefully and stop golimumab if the infection becomes serious.
- For patients who develop a systemic illness on golimumab, consider empiric anti-fungal therapy for those who reside in or travel to regions where mycoses, such as histoplasmosis, coccidioidomycosis or blastomycosis, are endemic.
- HBV many occur. Monitor HBV carriers during and several months after therapy. If reactivation occurs, stop golimumab and begin anti-viral therapy.
- The incidence of lymphoma was seen more often than in the general U.S. population. Cases of other malignancies have been observed among patients receiving a TNFi.
- Heart failure, worsening, or new onset, may occur. Stop golimumab if new or worsening symptoms occur.
- Demyelinating disease, exacerbation or new onset, may occur.
- Serious systemic hypersensitivity reactions, including anaphylaxis, may occur.
- Lupus-like syndrome—stop golimumab if the syndrome develops.
Dosage & Administration
Golimumab is administered by SQ injection. The recommended dose is 50 mg once a month in combination with MTX.
Commentary
The FDA approval of golimumab injection was based on the results of three multi-center, randomized, double-blind, controlled trials that enrolled more than 1,500 patients. The most common adverse reactions (>5%) were upper respiratory tract infection, nasopharyngitis and injection site reactions.
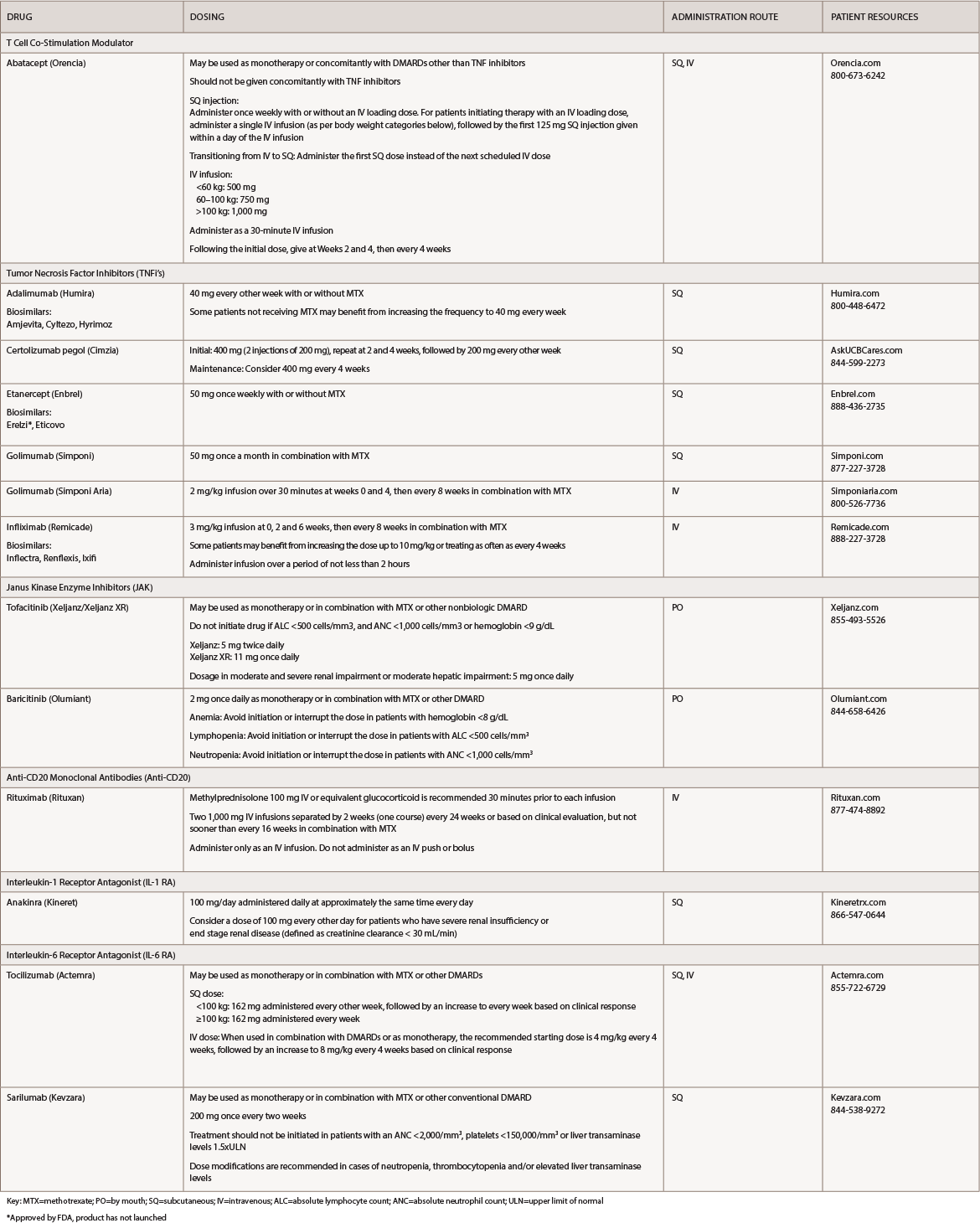
(click for larger image) Table 1: Rheumatoid Arthritis Medications at a Glance
Key: MTX=methotrexate; PO=by mouth; SQ=subcutaneous; IV=intravenous; ALC=absolute lymphocyte count; ANC=absolute neutrophil count; ULN=upper limit of normal
*Approved by FDA, product has not launched
Golimumab (Simponi Aria):18 infusion
Drug class: DMARD, TNFi
Boxed warning: Refer to *ISI (p. 14)
Warnings & Precautions: Refer to *Simponi
Dosage & Administration
Golimumab is administered by IV infusion. The recommended dose is 2 mg/kg over 30 minutes at Weeks 0 and 4, then every eight weeks in combination with MTX.
Commentary
The FDA approval of golimumab injection for IV use was based on results from a clinical trial that enrolled nearly 600 patients. The trial showed the drug significantly improved the signs and symptoms of RA, improved physical function and slowed the progression of structural damage. The most common adverse reactions (≥3%) were upper respiratory tract infection, increased alanine aminotransferase, viral infection, increased aspartate aminotransferase, decreased neutrophil count, bronchitis, hypertension and rash.
Infliximab (Remicade):19 infusion
Biosimilar(s): infliximab-dyyb (Inflectra),20 infliximab-abda (Renflexis),21 infliximab-qbtx (Ixifi)22
Drug class: DMARD, TNFi
Boxed warning: Refer to *ISI (p. 14) and
- Fatal hepatosplenic T cell lymphoma (HSTCL) have been reported in patients treated with TNFi’s, post-marketing. All infliximab cases occurred in IBD patients, who were mostly adolescent or young adult males. All had received azathioprine or 6-mercaptopurine concomitantly with infliximab at or prior to diagnosis.
Warnings & Precautions
- Do not give infliximab during an active infection. If an infection develops, monitor carefully and stop infliximab if the infection becomes serious.
- Invasive fungal infections—for patients who develop a systemic illness on infliximab, consider empiric anti-fungal therapy for those who reside in or travel to regions where mycoses, such as histoplasmosis, coccidioidomycosis or blastomycosis, are endemic.
- The incidence of malignancies, including lymphoma, was greater in infliximab-treated patients than in controls. Due to the risk of HSTCL, carefully assess the risks and benefits, especially in IBD patients, in men and in those receiving azathioprine or 6-mercaptopurine treatment.
- HBV reactivation can occur. Test for HBV infection before starting infliximab. Monitor HBV carriers during and several months after therapy. If reactivation occurs, stop infliximab and begin anti-viral therapy.
- Hepatotoxicity—rare severe hepatic reactions, some fatal or necessitating liver transplantation—has occurred Stop infliximab in cases of jaundice and/or marked liver enzyme elevations.
- Heart failure, new onset or worsening symptoms, may occur.
- Cytopenias—advise patients to seek immediate medical attention if signs and symptoms develop, and consider stopping infliximab.
- Hypersensitivity—serious infusion reactions, including anaphylaxis or serum sickness-like reactions, may occur.
- Demyelinating disease, exacerbation or new onset, may occur.
- Lupus-like syndrome—stop infliximab if syndrome develops.
- Live vaccines or therapeutic infectious agents should not be given with infliximab.
Dosage & Administration
Infliximab is administered by IV infusion over a period of not less than two hours. In conjunction with MTX, the recommended dose of infliximab is 3 mg/kg at Weeks 0, 2 and 6, then every eight weeks. Some patients may benefit from increasing the dose up to 10 mg/kg or treating as often as every 4 weeks.
Commentary
The FDA approved infliximab for treatment of RA based on data from a clinical trial that enrolled 428 patients at 34 clinical sites. At Week 30, 50% of all patients treated with infliximab plus MTX, compared with 20% of patients receiving MTX alone, achieved an ACR20 response. The most common adverse reactions (≥10%) were infections (e.g., upper respiratory, sinusitis, pharyngitis), infusion-related reactions, headache and abdominal pain.
Tofacitinib (Xeljanz/Xeljanz XR):23 tablets
Drug class: JAK inhibitor, DMARD
Boxed warning: Refer to *ISI (p. 14) and
- Lymphoma and other malignancies have been observed in tofacitinib-treated patients. Epstein-Barr virus-associated post-transplant lymphoproliferative disorder has been observed at an increased rate in renal transplant patients treated with tofacitinib and concomitant immunosuppressive medications.
- Compared with the 5 mg twice daily dose of tofacitinib or TNF blockers, the 10 mg dose has an increased risk of blood clots and death.24
Warnings & Precautions
- Avoid the use of tofacitinib during an active, serious infection, including localized infections.
- Use with caution in patients who may be at increased risk for gastrointestinal perforations.
- Monitor laboratory parameters: lymphocytes, neutrophils, hemoglobin, liver enzymes and lipids.
- Avoid the use of live vaccines with tofacitinib.
Dosage & Administration
Tofacitinib is available for oral administration. The dose of Xeljanz is 5 mg twice daily and the dose of Xeljanz XR is 11 mg once daily. It may be used as monotherapy or in combination with MTX or other nonbiologic DMARDs. Do not initiate the drug if the absolute lymphocyte count (ALC) is <500 cells/mm3, absolute neutrophil count (ANC) is <1,000 cells/mm3 or hemoglobin is <9 g/dL.
Commentary
Tofacitinib is the first drug of a new class of treatments called JAK inhibitors approved for the treatment of RA. The safety and efficacy of the drug were evaluated in seven clinical trials in adult patients with moderate to severe, active RA. In all of the trials, patients treated with tofacitinib saw a greater improvement in clinical response and physical functioning than patients treated with placebo. The most common adverse reactions (≥2%) were upper respiratory tract infection, nasopharyngitis, diarrhea and headache.
Baricitinib (Olumiant):25 tablets
Drug class: JAK inhibitor, DMARD
Boxed warning: Refer to *ISI (p. 14) and
- Lymphoma and other malignancies have been observed in baricitinib-treated patients.
- Thromboses, including deep venous thrombosis, pulmonary embolism and arterial thrombosis, some fatal, have occurred in patients treated with baricitinib. Venous thromboses have been reported more frequently in patients treated with 4 mg baricitnib.26
Warnings & Precautions
- Avoid the use of baricitinib during an active serious infection, including localized infections.
- Use with caution in patients who may be at increased risk for gastrointestinal perforations.
- Monitor laboratory parameters: lymphocytes, neutrophils, hemoglobin, liver enzymes and lipids, due to potential changes.
- Use with caution in patients who may be at increased risk for thrombosis.
- Avoid the use of live vaccines with baricitinib.
Dosage & Administration
Baricitinib is available for oral administration. The recommended dose
of baricitinib is 2 mg once daily. It may be used as monotherapy or in combination with MTX or other DMARDs. Do not initiate the drug if the ALC is <500 cells/mm3, ANC is <1,000 cells/mm3 or the hemoglobin is <8 g/dL.
Commentary
The FDA approved baricitinib based on the results of a clinical trial that included 527 patients who had an inadequate response or were intolerant to one or more TNF inhibitors. At 12 weeks, the researchers found 49% of patients treated with baricitinib achieved ACR20 response rates and improvement in all individual ACR20 component scores compared with 27% of patients in the placebo group. Patients in the baricitinib group showed significant improvements in physical function based on the Health Assessment Questionnaire Disability Index, with an average score of 1.71 prior to treatment and 1.31 at Week 12, compared with patients in the placebo group, who had an average score of 1.78 prior to treatment and 1.59 at Week 12. The most common adverse reactions (≥1) were upper respiratory tract infections, nausea, herpes simplex and herpes zoster.
Rituximab (Rituxan):27 infusion
Drug class: anti-CD20
Boxed Warning
- Fatal infusion-related reactions within 24 hours of rituximab infusion; approximately 80% of fatal reactions occurred with the first infusion. Monitor patients, and discontinue infusion for severe reactions.
- Severe mucocutaneous reactions have occurred, some with fatal outcomes.
- HBV reactivation, in some cases resulting in fulminant hepatitis, hepatic failure and death, has occurred.
- Progressive multifocal leukoencephalopathy (PML) resulting in death has occurred.
Warnings & Precautions
- Infections: Withhold rituximab, and institute appropriate anti-infective therapy.
- Cardiac adverse reactions: Discontinue infusions in case of serious or life-threatening events.
- Renal toxicity: Discontinue in patients with rising serum creatinine or oliguria.
- Bowel obstruction and perforation: Consider and evaluate for abdominal pain, vomiting or related symptoms.
- Immunizations: Live virus vaccinations prior to or during rituximab treatment are not recommended.
- Embryo-fetal toxicity: Can cause neonatal harm. Advise patients of the potential risk to neonates and use of effective contraception.
Dosage & Administration
Rituximab is administered only as an IV infusion. The dose of rituximab in combination with MTX is two 1,000 mg IV infusions separated by two weeks (one course) every 24 weeks or based on clinical evaluation, but not sooner than every 16 weeks. Administration of 100 mg of methylprednisolone via IV or equivalent glucocorticoid is recommended 30 minutes prior to each infusion.
Commentary
The FDA approved rituximab based on a clinical trial that enrolled 465 patients with moderate to severe RA. A total of 55% of the patients treated with the higher dose rituximab regimen showed a 20% or better improvement after six months, compared with 54% of patients on the low-dose regimen and 28% of patients taking a placebo. The most common adverse reactions (≥10%) were upper respiratory tract infections, nasopharyngitis, urinary tract infections, bronchitis, infusion-related reactions, serious infections and cardiovascular events.
Anakinra (Kineret):28 injection
Drug class: IL-1 RA, DMARD
Warnings & Precautions
- Discontinue use if serious infection develops.
- Avoid the use of anakinra in combination with TNF inhibitors.
- Hypersensitivity reactions, including anaphylactic reactions and angioedema, have been reported.
- The impact of treatment with anakinra on active and/or chronic infections and the development of malignancies is not known.
- Live vaccines should not be given concurrently with anakinra.
- Neutrophil counts should be assessed prior to initiating anakinra treatment, and while receiving anakinra, monthly for three months, and thereafter quarterly for a period up to one year.
Dosage & Administration
Anakinra is administered by SQ injection. The recommended dose of anakinra is 100 mg/day administered daily at approximately the same time each day. For RA patients who have severe renal insufficiency or end-stage renal disease (defined as creatinine clearance <30 mL/min), healthcare providers should consider a dose of 100 mg of anakinra administered every other day.
Commentary
Anakinra is the first selective antagonist of interleukin (IL) 1. In clinical trials, most patients experienced a decrease in inflammation and pain by Week 13 of treatment. Thirty-eight percent of patients treated with anakinra met ACR20 criteria for improvement compared with 22% of patients receiving placebo. The most common adverse reactions (≥5%) were injection site reaction, worsening of RA, upper respiratory tract infection, headache, nausea, diarrhea, sinusitis, arthralgia, flu like-symptoms and abdominal pain.
Tocilizumab (Actemra):29 injection, infusion
 Drug class: IL-6 RA, DMARD
Drug class: IL-6 RA, DMARD
Boxed Warning
- There is an increased risk of serious infections leading to hospitalization or death, including TB, bacterial, invasive fungal, viral and other opportunistic infections.
- If a serious infection develops, interrupt tocilizumab until the infection is controlled.
- Before starting treatment, perform a test for latent TB; if positive, start treatment for TB prior to starting the drug. Monitor all patients for development of active TB during treatment, even if the initial latent TB test was negative.
Warnings & Precautions
- Do not administer the drug during an active infection, including localized infections. If a serious infection develops, interrupt the drug until the infection is controlled.
- Gastrointestinal perforation—use with caution in patients who may be at increased risk.
- Laboratory monitoring is recommended due to potential consequences of treatment-related changes in neutrophils, platelets, lipids and liver function tests.
- Hypersensitivity reactions, including anaphylaxis and death, have occurred.
- Avoid the use of live vaccines with tocilizumab.
Dosage & Administration
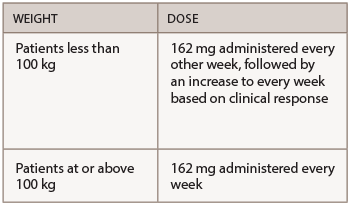
Tocilizumab is administered by SQ injection or IV infusion. It may be used as monotherapy or in combination with MTX or other DMARDs. The recommended SQ dose is based on the patient’s weight.
When used in combination with DMARDs or as monotherapy, the recommended initial IV infusion dose is 4 mg/kg every four weeks followed by an increase to 8 mg/kg every four weeks based on clinical response.
Commentary
Tocilizumab is the first IL-6 inhibitor approved for the treatment of RA. The FDA first approved the IV infusion formulation for adults with RA in January 2010 and the pre-filled syringe formulation for SQ injection for adults with RA in October 2013. The most common adverse reactions (>5%) were upper respiratory tract infections, nasopharyngitis, headache, hypertension, increased alanine transaminase (ALT) and injection site reactions.
Sarilumab (Kevzara):30 injection
Drug class: IL-6 RA, DMARD
Boxed Warning
- There is an increased risk of serious infections leading to hospitalization or death, including TB, bacterial, invasive fungal, viral, and other opportunistic infections in patients receiving sarilumab.
- If a serious infection develops, interrupt the drug until the infection is controlled.
- Before starting treatment, perform a test for latent TB; if positive, start treatment for TB prior to starting the drug. Monitor all patients for development of active TB during treatment, even if the initial latent TB test was negative.
Warnings & Precautions
- Do not administer the drug during an active infection, including localized infections. If a serious infection develops, interrupt the drug until the infection is controlled.
- Gastrointestinal perforation—risk may be increased with concurrent diverticulitis or concomitant use of non-steroidal anti-inflammatory drugs or corticosteroids. Promptly evaluate acute abdominal signs or symptoms.
- Laboratory monitoring is recommended due to the potential consequences of treatment-related changes in neutrophils, platelets,
lipids and liver function tests. - Hypersensitivity reactions, including anaphylaxis and death, have occurred.
- Avoid the use of live vaccines with sarilumab.
Dosage & Administration
The recommended dosage of sarilumab is 200 mg once every two weeks, administered as a SQ injection. It can be used as monotherapy or in combination with MTX or other conventional DMARDs. Treatment should not be initiated in patients with an ANC <2,000/mm3, platelets <150,000/mm3, or liver transaminase levels above 1.5 times the upper limit of normal. Dose modifications are recommended in cases of neutropenia, thrombocytopenia and/or elevated liver transaminase levels.
Commentary
The FDA approval for sarilumab was based on results from two clinical trials that enrolled more than 2,900 adults with moderate to severe, active RA who had inadequate response to previous treatment regimens. In both studies, the primary endpoint was improvement in signs and symptoms of RA, as measured by the proportion of patients who achieved a 20% improvement using the ACR criteria at Week 24. Both studies showed the sarilumab-treated group had an improvement in the primary endpoint and a greater improvement in physical function, compared with the placebo group. The most common adverse reactions (>3%) were neutropenia, increased ALT, injection site erythema, upper respiratory tract infections and urinary tract infections.
Mary Choy, PharmD, BCGP, FASHP, is a medical writer and editor living in New York City. Dr. Choy is director of pharmacy practice at the New York State Council of Health-system Pharmacists.
References
- Singh JA, Saag KG, Bridges L, et al. 2015 American College of Rheumatology Guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol. 2016 Jan;68(1):1–26.
- Kim Y, Oh H, Park, J, et al. Diagnosis and treatment of inflammatory joint disease. Hip Pelvis. 2017 Dec;29(4):211–222.
- van Vilsteren MV, Boot CR, Knol DL, et al. Productivity at work and quality of life in patients with rheumatoid arthritis. BMC Musculoskelet Disord. 2015 May;16:107.
- van der Heijde DM, van Leeuwen MA, van Riel PL, et al. Biannual radiographic assessments of hands and feet in a three-year prospective followup of patients with early rheumatoid arthritis. Arthritis Rheum. 1992 Jan:35(1):26–34.
- Fuchs HA, Kaye JJ, Callahan LF, et al. Evidence of significant radiographic damage in rheumatoid arthritis within the first 2 years of disease. J Rheumatol. 1989 May;16(5):585–591.
- Burmester GR, Pope JE. Novel treatment strategies in rheumatoid arthritis. Lancet. 2017 June;389:2338–2348.
- Rheumatoid Arthritis Support Network. Rheumatoid arthritis resources.
- FDA website. Orencia prescribing information. 2017 Dec.
- FDA website. Humira prescribing information. 2018 Dec.
- FDA website. Amjevita prescribing information. 2018 Mar.
- FDA website. Cyltezo prescribing information. 2017 Aug.
- FDA website. Hyrimoz prescribing information. 2018 Oct.
- FDA website. Cimzia prescribing information. 2019 Feb.
- FDA website. Enbrel prescribing information. 2018 May.
- FDA website. Erelzi prescribing information. 2016 Aug.
- FDA website. Eticovo prescribing information. 2019 Apr.
- FDA website. Simponi prescribing information. 2011 Dec.
- FDA website. Simponi Aria prescribing information. 2018 Feb.
- FDA website. Remicade prescribing information. 2018 Jun.
- FDA website. Inflectra prescribing information. 2018 Jul.
- FDA website. Renflexis prescribing information. 2019 Mar.
- FDA website. Ixifi prescribing information. 2017 Dec.
- FDA website. Xeljanz prescribing information. 2018 May.
- Xeljanz label. Boxed warning (added July 2019). Pfizer Laboratories a division of Pfizer Inc.
- FDA website. Olumiant prescribing information. 2018 May.
- Olumiant. Select important safety information: Warning related to serious infections, malignancy, and thrombosis.
- FDA website. Rituximab prescribing information. 2019 Jan.
- FDA website. Kineret prescribing information. 2018 Jun.
- FDA website. Actemra prescribing information. 2019 Apr.
- FDA website. Kevzara prescribing information. 2018 Apr.