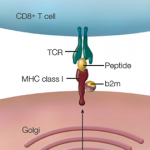
At its most basic level, celiac disease begins as an immune response to wheat proteins that then expands into an autoimmune disease with both gastrointestinal and neurological implications. Celiac disease is further complicated by the fact that it is multifactorial and polygenic. Fifty years ago, scientists identified the major histocompatibility complex (MHC) as a risk factor for celiac disease, with the primary risk factor being MHC Class II alleles encoding HLA-DQ2.5, HLA-DQ8 and HLA-DQ2.2. More recently, genome-wide association studies have identified 42 additional loci that contribute to celiac disease susceptibility.
At its most basic level, celiac disease begins as an immune response to wheat proteins that then expands into an autoimmune disease with both gastrointestinal and neurological implications. Celiac disease is further complicated by the fact that it is multifactorial and polygenic. Fifty years ago, scientists identified the major histocompatibility complex (MHC) as a risk factor for celiac disease, with the primary risk factor being MHC Class II alleles encoding HLA-DQ2.5, HLA-DQ8 and HLA-DQ2.2. More recently, genome-wide association studies have identified 42 additional loci that contribute to celiac disease susceptibility.
Bana Jabri, MD, PhD, professor of medicine at the University of Chicago, and Ludvig M. Sollid, MD, PhD, professor at the Center for Immune Regulation at the University of Oslo in Norway, detailed the immunological research that has been conducted to date to elucidate celiac disease and published their review online April 15 in the Journal of Immunology. They describe celiac disease as an autoimmune disease with CD4 T cells, autoantibodies and effector intraepithelial cytotoxic T lymphocytes (IE-CTL).1