Study Findings
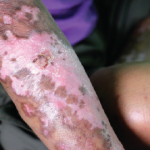
Using multiple lines of evidence, the researchers assessed the role of DPP4 expression on fibrosis in systemic sclerosis. They used skin biopsy samples to isolate dermal fibroblasts from 23 people with systemic sclerosis, matched with 21 healthy controls. In vivo studies included three classic mouse models used to mimic the symptomatology of systemic sclerosis. These were bleomycin-induced skin and lung fibrosis, sclerodermatous chronic graft-versus-host disease, and the tight skin mouse model (fibrillin-1 mutation mouse).1,3
To help clarify the role of DPP4, the team employed a strain of mice genetically bred to be unable to express DPP4—DPP4 knockouts—as well as a control strain (wild type). This helped the team tease apart the role of DPP4, as they saw how the different mice responded to induction of the systemic sclerosis model. They also explored sitagliptin, a DPP4 inhibitor, to see how its usage affected induced systemic sclerosis in these various mouse models.1
Dr. Soare and colleagues found the expression of DPP4 was increased in patients with systemic sclerosis compared to controls, with fibroblasts as the dominant cell type expressing DPP4. For example, almost 76% of the fibroblasts of systemic sclerosis patients expressed DPP4, compared to about 29% of fibroblasts in people with healthy skin. This pattern also held in mouse models of the disease. The population of fibroblasts expressing DPP4 seemed to mark a subset of highly activated fibroblasts, which expressed higher levels of collagen and of genetic markers for myofibroblasts. However, serum levels of DPP4 did not differ, consistent with earlier work, suggesting DPP4 is locally regulated.1
Moreover, this expression of DPP4 appeared to increase in proportion to TGF‑β. Overexpression of DPP4 promoted activation of fibroblasts. Conversely, Dr. Soare notes, “Inactivation of DPP4 blocked TGF-β-induced fibroblast-to-myofibroblast differentiation and reduced the release of collagen in vitro.”
She adds, “Genetic or pharmacologic inhibition of DPP4 also ameliorated experimental dermal and pulmonary fibrosis induced by bleomycin or by sclerodermatous processes [chronic graft-vs.-host disease].” Significantly, not only did pharmacological inhibition of DPP4 prevent further fibrosis progression, it also induced regression of existing fibrosis to below pre-treatment levels.1
Moreover, in addition to the direct effect on fibroblasts, inhibition of DPP4 seemed to also reduce inflammation.1 Dr. Soare explains, “In murine models of systemic sclerosis, treatment with DPP4 inhibitors reduced leukocyte counts and in particular T cell and B cell infiltration, both of which are centrally involved in the pathogenesis of the disease.”
Through molecular analysis, the team demonstrated the importance of the intracellular signaling mediator ERK (extracellular signal-regulated kinase), whose inhibition has previously been shown to ameliorate fibrosis in experimental models.5 Dr. Soare remarks, “Inhibition of the non-canonical TGF-β signaling mediator, ERK, inhibits the stimulatory effects of TGF-β on DPP4 expression.” Moreover, treatment of incubated human fibroblasts with DPP4 inhibitors prevented TGF-β from having its normal stimulatory effects on ERK signaling. Dr. Soare reports that other known intracellular cascades regulated by TGF-β were not found to be affected by inhibition of DPP4.1