
There is a striking association of sJIA with secondary hemophagocytic lymphohistiocytosis, also known as macrophage activation syndrome (MAS). Although approximately 10% of sJIA patients develop overt MAS, up to 30% of children have evidence of subclinical MAS, characterized by a number of biochemical and immunological markers of MAS, including elevated levels of soluble CD163.2 In fact, some markers of MAS such as elevations of serum ferritin and d-dimers are considered typical laboratory manifestations of active systemic disease. It is interesting to note that both active sJIA and MAS are characterized by natural killer (NK) cell dysfunction. Full-blown MAS complicating sJIA is associated with significant morbidity and potentially mortality.
The following case represents a typical patient with early sJIA. We will present the case and discuss the treatment choices and outcome.
The Case
A 14-year-old boy was seen in the rheumatology clinic, having been unwell for approximately two months. He initially presented with diffuse arthralgias but subsequently developed fevers up to 102°F, usually once daily in the evening or early morning. He missed several days of school each week and estimated that he had lost 4 to 5 kg in weight. On physical examination, he had a typical systemic rash, diffuse lymphadenopathy, and oligoarthritis involving both wrists and one elbow. He did not have evidence of serositis or hepatosplenomegaly. After investigations excluded infections, malignancies, and other rheumatic diseases, a diagnosis of sJIA was made and he was treated with naproxen 500 mg bid. There was no improvement in his symptoms or laboratory markers when he was seen for a follow-up visit (see Table 2). Once the patient failed this initial nonsteroidal antiinflammatory drug (NSAID) trial, a decision had to be made about the next step in treatment.
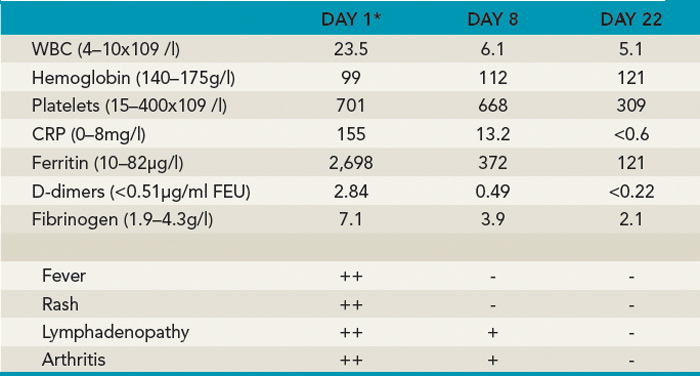
Key: ++=severe, +=moderate, -=absent *Baseline before treatment with anakinra.
Typical Disease Progression
Despite the protean presenting manifestations of the disease, up to 40% of children have a good long-term outcome with a monocyclic course that enters a permanent remission after a variable number of months. A small number of patients have a polycyclic course with recurrent episodes of active disease and periods of true remission, while more than half of the patients have a more severe, persistent disease course. Prognostic markers of poor outcome have been identified, but do not have optimum sensitivity or specificity (see Table 3).3,4 Among the group with persistent disease, the most severely affected children have ongoing systemic symptoms, early destructive polyarthritis, growth failure, and all the attendant side effects of chronic systemic corticosteroid treatment, on which children are often dependent to maintain a reasonable level of physical activity and quality of life. Traditional disease-modifying antirheumatic drugs (DMARDs), such as methotrexate, have limited efficacy for the joint disease and virtually no impact on the systemic features of the disease.

In addition to the disease-related complications in the acute phase, such as serositis, severe anemia, and MAS, and the devastating consequences of the polyarthritis of sJIA, treatment-related complications have resulted in major morbidity in this population. Until recently, many patients required relatively large doses of corticosteroids to control systemic disease, articular disease, or both. In fact, in some patients, treatment-related morbidity has been worse than disease-related morbidity. This has been observed in two particular areas where steroid treatment has been additive to disease-related manifestations. Growth delay is commonly observed and is certainly more prevalent in patients with worse disease requiring more treatment. Several studies have reported the use of growth hormone (GH) resulting in improved linear growth.5 However, there is still controversy regarding whether these children have an improved final height or whether their growth is only accelerated during the time that the GH is being administered.6 Secondly, steroid- and disease-related osteopenia and osteoporosis can have serious consequences, including pathologic fractures; if these involve the vertebral bodies, final height may also be impacted. The pain from the fractures also results in immobility, further accelerating the development of osteopenia. Medications other than corticosteroids are needed to reduce—or, in fact, prevent—these severe complications.
Treatment Advances
Advances in the understanding of the disease pathogenesis have resulted in the application of biologic treatments directed at inhibiting cytokines associated with the disease, particularly interleukin (IL) 1 and IL-6. Recent studies suggest that targeting these specific cytokines may result in much better outcomes than treating with corticosteroids and traditional DMARDs and may avoid the devastating toxicity of high-dose corticosteroid treatment.
Many investigators consider sJIA an autoinflammatory disease since it is not strongly associated with human leukocyte antigen (HLA) alleles, autoantibodies, or autoreactive T cells, but instead it is considered to involve dysregulation of the innate immune system. Although levels of circulating IL-1β are low or undetectable, Pascual and colleagues have provided convincing evidence for the role of IL-1β in the disease pathogenesis by demonstrating increased expression of IL-1–related genes in sJIA and showing that serum from sJIA patients induces production of IL-1β in peripheral blood mononuclear cells from healthy donors.7
Uncontrolled, open-label studies have shown that anakinra, an IL-1 receptor antagonist, improves systemic symptoms and laboratory features of inflammation in the majority of patients, but generally less impressive improvement is seen in the arthritis. An important study by Gattorno and colleagues identified two groups of patients: good responders, whose arthritis improved rapidly within the first week of treatment, and incomplete or nonresponders, whose arthritis either did not improve significantly or improved transiently with early recurrence.8 There is only one published controlled clinical trial of anakinra, the ANAJIS trial, in which 24 patients with sJIA that was steroid dependent were randomized to receive anakinra 1 mg/kg/day by subcutaneous injection or placebo.9 Eight of 12 patients (67%) who received anakinra met the primary outcome of a modified pediatric ACR 30 response after one month, compared with only one of 12 patients (8%) who received placebo. Modified pediatric ACR 50 and pediatric ACR 70 responses were achieved by 58% and 42% of patients, respectively.
After one month, 10 of the patients who had been treated with placebo and were considered nonresponders received anakinra and achieved a modified pediatric ACR 30 response after one month of treatment. However, at the end of the second month of treatment with anakinra, seven of the eight original responders at month one became nonresponders. Of the entire study group of 24 patients, only six out of 17 (35%) who received anakinra were considered responders after six months. After one year of treatment with anakinra, seven out of 16 (44%) were considered responders. Six of these patients had inactive disease and had been completely weaned off systemic corticosteroids.
It has been suggested that initiation of anakinra early in the disease course may improve outcome. In a report of 16 corticosteroid-naive patients with sJIA, 14 out of 16 responded to anakinra, the majority of patients achieving a pediatric ACR 90 response within three weeks of treatment.10 Another series reported the use of anakinra as part of the initial DMARD regimen in 46 patients with sJIA.11 Two-thirds of patients were on systemic corticosteroids, with or without other DMARDs. Fifty-nine percent had an excellent (“complete”) response, with almost all patients showing rapid improvements in fever and rash. There was a slower response of the arthritis to treatment, with persistent synovitis in 39% of patients at 30 days, 27% of patients at three months, and 15% of patients at six months. Inflammatory markers normalized in most patients with one month. Interestingly, 10 of the 46 patients received anakinra without steroids or other DMARDs.

Overall, anakinra appears to be effective in a high proportion of sJIA patients, with improvements in both systemic symptoms and arthritis in approximately 50%. However, some responders may subsequently have disease flares. Doses >1.5 mg/kg/day are likely to be more effective. Although data are limited, suggested predictors of response to anakinra include lower number of inflamed joints, higher neutrophil counts, involvement of predominantly large joints, and age seven years or older at disease onset.8,11,12
There are currently no published controlled studies of other IL-1 inhibitors in sJIA. Preliminary reports suggest that longer-acting agents such as rilonacept and canakinumab are effective in treating both systemic symptoms and arthritis in steroid-dependent sJIA.13,14
SJIA is also characterized by markedly elevated levels of IL-6 in serum and synovial fluid. Moreover, elevated IL-6 serum levels have been shown to correlate with the fever spikes and are associated with the development of arthritis, hypochromic microcytic anemia, high inflammatory markers, growth failure, and osteoporosis seen in this disease.15 Tocilizumab, a humanized monoclonal antibody that binds both soluble and membrane-bound IL-6 receptors, showed excellent results in initial open-label trials of sJIA. A Japanese controlled trial of tocilizumab in sJIA adopted a randomized withdrawal design.16 In the first open-label phase, all patients whose disease was not adequately controlled by systemic corticosteroids and who had an elevated CRP received tocilizumab 8 mg/kg/day every other week by infusion. After six weeks, 91%, 86%, and 68% achieved modified pediatric ACR 30, 50, and 70 responses, respectively. All responders were subsequently randomized to receive tocilizumab or placebo. There was a much higher disease-flare rate in the placebo group.
In a large prospective placebo-controlled trial in active sJIA, patients were randomized to receive tocilizumab or placebo in a 2:1 ratio.17 Those weighing less than 30 kg received tocilizumab 12 mg/kg every two weeks, and those weighing more than 30 kg received 8 mg/kg. At 12 weeks after randomization, modified pediatric ACR 30, 50, 70, and 90 responses were achieved by 90.7% (vs 24.3%), 85.3% (vs 10.8 %), 70.7% (vs 8.1%) and 37.3% (vs 5.4%), respectively. Fever was seen in 14.7% of patients on tocilizumab (vs 79.2% on placebo) and rash was seen in 36.4% of tocilizumab-treated patients (vs 88.9% of patients on placebo). All patients were eligible to receive tocilizumab after the 12-week placebo-controlled phase. After one year of treatment, more than 50% of patients had discontinued corticosteroids, 49% had no active arthritis, 60% had achieved an ACR 90 response, and 28% met criteria for inactive disease.
Treatment Recommendations
Given the efficacy of IL-1 and IL-6 inhibition for active sJIA, and considering the well-known and prominent side effects of prolonged systemic corticosteroids, what is the role of corticosteroids and these biologic agents in the treatment of this disease? In 2011, the ACR recommendations for the treatment of JIA were published.18 However, these recommendations were based on limited available data regarding the use of anakinra in sJIA and did not consider the use of longer-acting IL-1 inhibitors or tocilizumab in sJIA.
The ACR recommendations suggest that there is a role for a trial of NSAIDs for up to two weeks if disease activity is relatively low and poor prognostic features are absent. Patients with high disease activity (such as symptomatic pericarditis) are recommended to receive systemic corticosteroids and if they prove unresponsive, anakinra is recommended. However, considering the available evidence and expert opinion, the recommendations do recognize that anakinra could be used as a first-line treatment for active systemic symptoms of sJIA, particularly if there is persistent fever and if poor prognostic features are present.
In view of the more recent data alluded to above, one might also consider tocilizumab as an alternative to IL-1 inhibitors. The ACR recommendations for children who have active arthritis but no systemic features include NSAIDS, followed by methotrexate. If there is no response to methotrexate, the recommendations suggest that patients should receive either anakinra or a tumor necrosis factor–α inhibitor. The potential role of IL-1 and IL-6 inhibitors for arthritis without systemic features remains to be clarified.
Back to the Case
The patient was treated with anakinra 100mg (2 mg/kg) daily by subcutaneous injection. There was a dramatic improvement in his clinical and laboratory features after less than one week of treatment. Similar improvements might be anticipated with the use of high-dose systemic corticosteroids or tocilizumab.
Although clinical trials have addressed the use of anakinra or tocilizumab in patients who have not responded adequately to systemic corticosteroids, there is increasing use of these agents in patients with active systemic features and arthritis, even prior to receiving corticosteroids. The role of these agents in corticosteroid-naive patients requires further study. The results of a proposed prospective study of the comparative effectiveness of these agents in early sJIA may help to elucidate the most appropriate use of each of these agents. In the interim, systemic corticosteroids still have an important role in treating acute severe manifestations of the disease and severe complications such as symptomatic pericarditis and in treating MAS.
Disclosure
Dr. Schneider is a consultant for Hoffmann-La Roche.
Dr. Schneider is associate chair (education) in the division of rheumatology and associate professor in the department of paediatrics and a rheumatologist in the division of rheumatology at the University of Toronto and The Hospital for Sick Children in Ontario, Canada. Dr. Laxer is a rheumatologist in the division of rheumatology at The Hospital for Sick Children and professor of paediatrics and medicine at the University of Toronto.
References
- Petty, RE, Southwood TR, Manners P, et al. International league of associations for rheumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton 2001. J Rheumatol. 2004; 31:390-392.
- Grom AA, Mellins ED. Macrophage activation syndrome: Advances towards understanding pathogenesis. Curr Opin Rheumatol. 2010;22:561-566.
- Spiegel LR, Schneider R, Lang BA, et al. Early predictors of poor functional outcome in systemic-onset juvenile rheumatoid arthritis: A multicenter cohort study. Arthritis Rheum. 2000;43:2402-2409.
- Modesto C, Woo P, García-Consuegra J, Merino R, García-Granero M, Arnal C, Prieur AM. Systemic onset juvenile chronic arthritis, polyarticular pattern and hip involvement as markers for a bad prognosis. Clin Exp Rheumatol. 2001;19:211-217.
- Bechtold S, Ripperger P, Dalla Pozza R, Bonfig W, Häfner R, Michels H, Schwarz HP. Growth hormone increases final height in patients with juvenile idiopathic arthritis: Data from a randomized controlled study. J Clin Endocrinol Metab. 2007; 92:3013-3018.
- Al-Mutair A, Bahabri S, Al-Mayouf S, Al-Ashwal A. Efficacy of recombinant human growth hormone in children with juvenile rheumatoid arthritis and growth failure. J Pediatr Endocrinol Metab. 2000;13:899-905.
- Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201:1479-1486.
- Gattorno M, Piccini A, Lasigliè D, et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008;58:1505-1515.
- Quartier P, Allantaz F, Cimaz R, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis. 2011;70:747-754.
- De Jager W, Vastert SJ, Noordman BJ, Holzinger D, Kuis W, Prakken B, Wulffraat N. Anakinra restores the defective IL-18 NK cell axis in steroid naïve systemic onset JIA patients. Ann Rheum Dis. 2011;70(S3)89.
- Nigrovic PA, Mannion M, Prince FH, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis. Arthritis Rheum. 2011;63:545-555.
- Zeft A, Hollister R, LaFleur B, et al. Anakinra for systemic juvenile arthritis: The Rocky Mountain experience. J Clin Rheumatol. 2009;15:161-164.
- Lovell, DJ, Giannini EH, Kimura Y, et al. Long-term safety and efficacy of rilonacept in patients with systemic juvenile idiopathic arthritis (SJIA). Arthritis Rheum. 2009;60:S768.
- Ruperto N, Quartier P, Wulffraat N, et al. A phase II, multicenter, open-label study evaluating dosing and preliminary safety and efficacy of canakinumab in systemic juvenile idiopathic arthritis with active systemic features. Arthritis Rheum. 2012;64:557-567.
- de Benedetti F, Martini A. Targeting the interleukin-6 receptor: A new treatment for systemic juvenile idiopathic arthritis? Arthritis Rheum. 2005;52:687-693.
- Yokota S, Imagawa T, Mori M, et al. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: A randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet. 2008;22:998-1006.
- de Benedetti F, Brunner H, Ruperto N, et al. Efficacy and safety of tocilizumab in patients with systemic juvenile idiopathic arthritis: 2-Year data from a phase III trial. Arthritis Rheum. 2011;63:4047.
- Beukelman T, Patkar NM, Saag KG, et al. 2011 American College of Rheumatology recommendations for the treatment of juvenile idiopathic arthritis: Initiation and safety monitoring of therapeutic agents for the treatment of arthritis and systemic features. Arthritis Care Res (Hoboken). 2011; 63:465-482.

