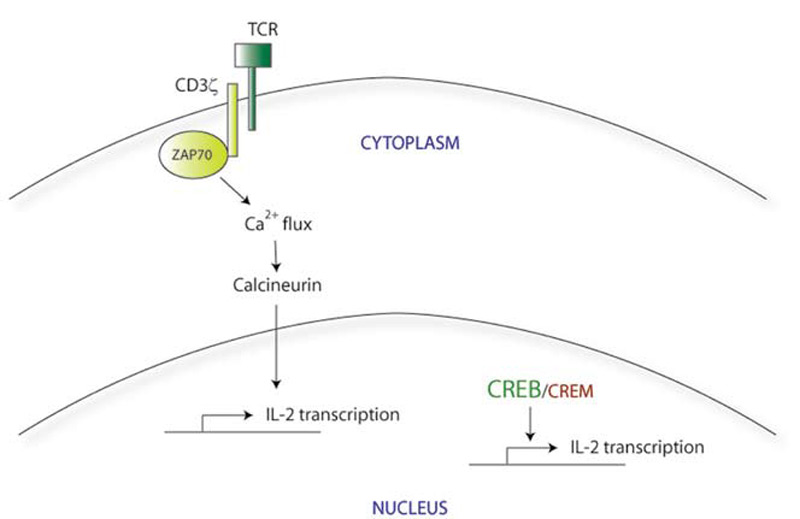
T-Cell Signaling Abnormalities: TCR Signaling
Normally, initial binding of the TCR to its cognate foreign antigen on the surface of APCs triggers the clustering of other TCR molecules together at the cell surface, along with their downstream signaling components. The CD3 molecules, which include several different transmembrane proteins, are part of this signaling complex. CD3ζ in particular activates the intracellular protein zeta-associated protein 70 (ZAP70), which further transmits the activation signal leading to calcium influx and activation of transcription factors via calcineurin. This in turn leads to rapid upregulation of proinflammatory genes, including IL-2. (See Figure 1A)
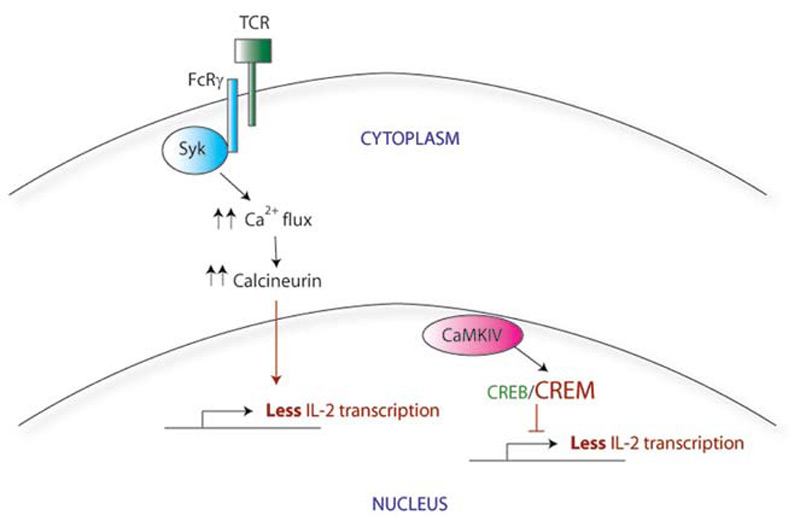
Calcium flux after activation of the TCR in SLE T cells is faster and stronger compared with that in normal T cells. This increased TCR response is probably multifactorial in origin, although one key contributor is the swapping out of one of the TCR subunits. SLE T cells have lower levels of CD3ζ than in normal cells and instead express a homologous protein that normally plays a role in other signaling pathways. The Fcε receptor common gamma chain (FcRγ), a component of the IgE receptor, is able to assemble with the TCR complex, substituting for the missing CD3ζ.1 Although similar in structure to CD3ζ, FcRγ signals through a different adaptor protein, Syk, rather than ZAP70. (See Figure 1B) The correlation between this substitution and the faster calcium flux response seen in SLE T cells is demonstrated by two experiments: first, forced expression of the FcRγ in normal T cells also increases the calcium influx response; and second, a Syk inhibitor normalizes the signal in SLE T cells.
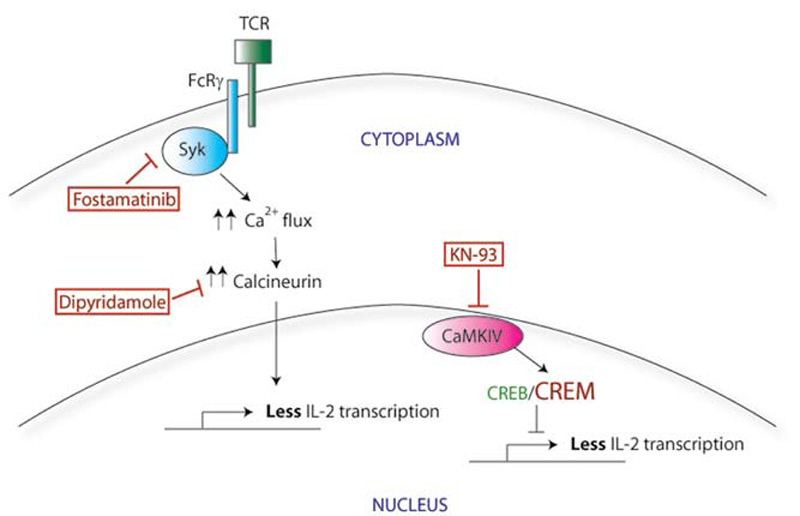
The mechanism by which this faster and stronger signaling response in SLE T cells results in less IL-2 production remains unclear. However, two new therapies targeting this signaling response may prove useful therapeutically (see Figure 1C). Calcium influx can be blocked through use of calcineurin inhibitors such as cyclosporine or tacrolimus, although the renal toxicity of these medications precludes widespread use in SLE. Recently, dipyridamole, a commonly used antiplatelet agent, has been shown to inhibit calcineurin and calcium flux signaling in T cells. Dipyridamole was further able to delay disease The mechanism by which this faster and stronger signaling response in SLE T cells results in less IL-2 production remains unclear. However, two new therapies targeting this signaling response may prove useful therapeutically (see Figure 1C, p. 24). Calcium influx can be blocked through use of calcineurin inhibitors such as cyclosporine or tacrolimus, although the renal toxicity of these medications precludes widespread use in SLE. Recently, dipyridamole, a commonly used antiplatelet agent, has been shown to inhibit calcineurin and calcium flux signaling in T cells. Dipyridamole was further able to delay disease progression in a mouse model of lupus.2 Second, since a Syk inhibitor was also able to normalize calcium signaling in vitro, this effect was further examined in vivo. Fittingly, fostamatinib, a small -molecule inhibitor of Syk, attenuates the disease phenotype in lupus-prone mice.3 It remains to be seen whether dipyridamole will have therapeutic potential in humans with SLE. On the other hand, fostamatinib is soon to enter the market for the treatment of rheumatoid arthritis, and we anticipate that there will be additional studies of fostamatinib in SLE as well.
T-Cell Signaling Abnormalities: Gene Expression
There appear to be many explanations for why T cells from SLE patients express lower levels of CD3ζ. These include decreased transcription, decreased mRNA stability, alternative splicing, and increased protein degradation. For example, the CD3ζ gene promoter contains a cyclic AMP response element (CRE). The CRE-modulator (CREM) transcription factor binds to this element, repressing CD3ζ transcription. The IL-2 gene promoter also contains a CRE site; CREM represses IL-2 transcription in a similar manner. In contrast, the CRE-binding protein (CREB) also binds to the same CRE site, but acts as an activator of IL-2 transcription. SLE T cells show simultaneously increased CREM expression and decreased CREB activity.4 Taking together data from these experiments, it appears that this CREB/CREM imbalance in SLE T cells at least partially explains the decreased CD3ζ and IL-2 expression in these cells.
Correcting the CREB/CREM imbalance (and by translation, the defect in CD3ζ and IL-2) opens up a new area for therapeutic intervention. CREM expression is regulated in part by a calcium-responsive kinase, the calmodulin-dependent kinase IV (CaMK IV), and SLE patients show higher levels of CaMK IV in the nucleus. Interestingly, KN-93, a small molecule inhibitor of CaMK IV, has shown efficacy in treating mouse models of lupus, suggesting a promising area for investigation of new therapy.5
Functional Abnormalities of T Cells in SLE
Similar to the situation with T-cell signaling, studies have identified a variety of defects in T-cell function in SLE. For example, cytotoxic activity of CD8 T cells from SLE patients is diminished.6 An inability to suppress infected cells may lead to ongoing polyclonal B-cell activation and autoantibody production; furthermore, a defect in cell killing may also explain the predisposition of SLE patients towards macrophage activation syndrome. The potential consequences of decreased IL-2 production in SLE are not completely known but are likely to be many, given the many functions of IL-2 in regulating immune responses.
Regulatory Activity
Regulatory T cells (Treg) are a T-cell subset that can suppress the activity of other T cells, self-reactive T cells in particular. SLE patients have been described to have lower Treg numbers in a manner correlating with disease activity.7 Treg survival and function is dependent on IL-2, as demonstrated by the fact that mice that lack IL-2 are also deficient in Treg cells. The relative deficiency in IL-2 observed in SLE patients, therefore, may contribute to lower Treg numbers and thus increased disease activity.
Theoretically, autologous Treg cells could be expanded ex vivo and re-infused into a patient, potentially correcting the regulatory cell defect. This is obviously a complicated and technically difficult proposition; however, this approach has been used with some success in mouse models of lupus.8
B-Cell Help
The production of antigen-specific antibodies is dependent on the activation and differentiation of B cells; this stimulation is, in turn, dependent on B-cell interaction with T cells. The main costimulatory signal is provided by the binding of CD40L on the surface of T cells with CD40 on B cells. SLE T cells express higher levels of CD40L, which may have a hyperstimulatory effect on B cells.9 Attempts have been made to develop treatments that inhibit CD40:CD40L interactions and thereby hinder B-cell stimulation. An anti-CD40L antibody has been tested in patients with lupus. Initial results from phase I/II trials were promising, with decreased anti-dsDNA titers and increased complement levels. Unfortunately, an increase in thromboembolic events was also seen.10 The reason for thromboembolism is not certain. Normal platelets also express CD40L, and, while not completely elucidated yet, anti-CD40L treatment may have activating effects on platelets.
Migration and Adhesion
T cells display various chemokine receptors that allow them to respond to inflammatory signals and migrate out of the circulation into the tissue. SLE T cells show altered migratory activity in states of inflammation. For example, the migration of cells in response to CXCL12 (a chemokine produced by multiple tissue cell types) is faster in SLE T cells.11 This may be due to increased expression or activation of CXCR4, the receptor for CXCL12. In addition, kidneys from patients with lupus nephritis show high levels of CXCL12, further supporting the role of the CXCR4/CXCL12 axis in T-cell homing to the kidney. Of note, B cells from SLE patients also have higher CXCR4 levels, which has been shown to correlate with disease activity, renal involvement, and neuropsychiatric disease.12 Blocking the CXCR4/CXCL12 axis represents another possible therapeutic approach for SLE treatment, and this has already shown some promise in mouse models of disease.13 Intriguingly, this approach is also being considered as potential cancer treatment, as the CXCR4/CXCL12 axis has been implicated in mediating cancer metastasis.
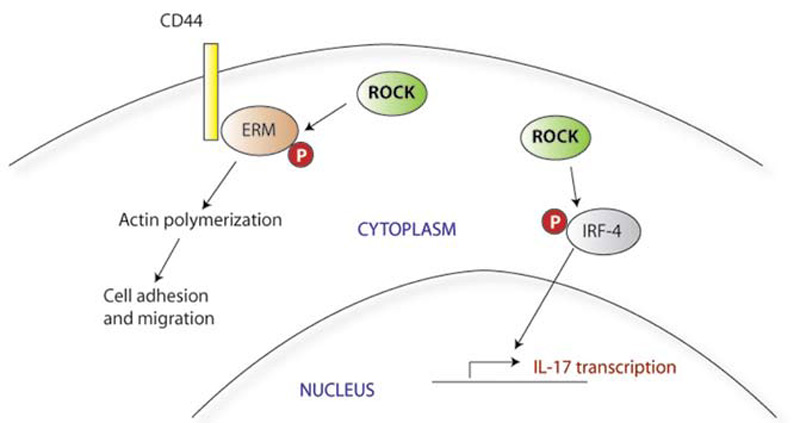
SLE T cells express higher levels of CD44, a surface adhesion molecule, and in vitro show stronger adhesion and faster migration in response to chemokines. CD44 occurs as a number of different isoforms; the distinct functions and characteristics of these variants are unclear. SLE T cells express higher levels of the CD44v3 and CD44v6 in particular, and this pattern of adhesion molecule expression is also seen in T cells infiltrating the kidneys of patients with lupus nephritis.14 CD44v3 (but not CD44v6) also has been implicated in the T-cell infiltrates of autoimmune skin conditions, including those associated with lupus. Within the cell, CD44 signals through its association with ezrin, radixin, and moesin (collectively known as ERM). Upon activation of CD44, the ERM signaling molecules are phosphorylated by rho kinase (ROCK); ERM proteins show higher levels of phosphorylation in patients with lupus.11 Suppression of the CD44 activation pathway by silencing CD44 expression or inhibiting ROCK normalizes the enhanced migratory responses of SLE T cells and may therefore have therapeutic applications. ROCK inhibitors have been shown to have other antiinflammatory effects as well, including inhibition of IL-17 and other inflammatory cytokines in mouse models of autoimmunity.15 (See Figure 2)
Th17 and Double-Negative T Cells
Th17 cells are a subset of T helper cells that produce IL-17. This cell type has been shown to play a role in a number of autoimmune diseases, including multiple sclerosis and inflammatory bowel disease. T cells secreting IL-17 have also been found in increased numbers in patients with SLE, including in the kidneys of patients with lupus nephritis. The production of IL-17 is thought to amplify the local inflammatory response by stimulating the recruitment other immune effector cells. Antibodies inhibiting the IL-17 pathway are already being evaluated for the treatment of rheumatoid arthritis; it remains to be seen whether anti–IL-17 strategies will also be useful for treating SLE.16
Under normal circumstances, almost all circulating T cells will express either CD4 or CD8 surface markers. A subpopulation of CD4–, CD8–(double-negative, or DN) T cells have also been described, although their function is not completely clear. DN T cells appear to have regulatory properties as they can suppress other T-cell responses. However, the DN T-cell population is also expanded in some autoimmune conditions. In SLE, DN T cells play an inflammatory role and can be found producing IL-17 within the kidneys of patients with lupus nephritis.17 Local infiltration of T cells and other inflammatory cells into the kidneys suggests that secreted cytokines and chemokines may be detectable in the urine. A change in cytokine profile in the urine may precede the development of renal damage and could therefore serve as a biomarker predictive of renal involvement.18
Oxidative Stress
Mitochondrial dysfunction has also been described in SLE T cells. This dysfunction leads to the increased production of destructive reactive-oxygen intermediates and oxidative stress. The mechanisms and consequences of this abnormality are complex and not fully understood. SLE T cells show increased activity of mammalian target of rapamycin (mTOR), a kinase that regulates mitochondrial transmembrane potential. Among other effects, this increased activity results in abnormal calcium flux and contributes to decreased CD3ζ expression, the central signaling defect in SLE T cells. Treating SLE T cells with rapamycin, an mTOR inhibitor, restores CD3ζ expression and normalizes downstream signaling.19 Rapamycin also improves the disease phenotype and prolongs survival of lupus-prone mice. In humans, rapamycin treatment showed some benefit in the treatment of nine SLE patients with refractory disease, but more investigation will be needed to determine whether this will be useful for patients more broadly.20
Future Directions
The myriad T-cell abnormalities that have been described with SLE make for a confusing, complicated picture of immune dysregulation. When considered individually and in vitro, many of these abnormalities vary with disease activity or recapitulate certain aspects of the disease in vivo. The SLE patient population in practice, however, is a clinically and genetically heterogeneous group. There is no one single marker that is useful practically as a biomarker for this diverse population. Recently, our group has described the use of a gene expression array that can capture a broader picture of T-cell dysfunction. This is a single test that allows the simultaneous measurement of expression levels for a panel of genes. We selected 30 genes previously reported to have aberrant expression or function in SLE, including CREM, Syk, CD3ζ, and other factors mentioned in this review. Analysis of gene-expression patterns showed that the SLE samples segregated from normal controls.21 Differential patterns of expression also emerged that were specific for the type of organ system involvement. As therapies targeting various aspects of T-cell dysfunction come into clinical use, these gene expression patterns could also be used to predict responses to specific treatments. (See Figure 3)
New research into epigenetic regulation of gene expression also may provide further insight into how environmental factors may come into play in the pathogenesis of autoimmunity. Several studies of DNA methylation in T cells have shown significant differences in methylation patterns between SLE T cells compared with normal controls.22,23 In general, SLE T cells show lower methylation levels for many genes, including some previously implicated in SLE pathogenesis. As an example, our group has shown that increased expression of protein phosphatase 2A (PP2A, an enzyme thought to contribute to the decreased IL-2 phenotype in SLE), is due to hypomethylation of its promoter.24 Less is known about histone modification in autoimmunity, although this is also an area of active research. Histone deacetylase inhibitors are already in development for treatment of certain cancers, and there are promising studies of these compounds in mouse models of lupus.25
As technology continues to advance, it may soon be possible to measure methylation patterns and other gene expression profiles easily, perhaps even as a bedside point-of-care test. Such a profile could be readily applied in clinical practice as a biomarker to predict disease activity, organ system involvement, and response to targeted therapy. Small-molecule inhibitors that target specific aberrations of gene expression in SLE could then be selectively used depending on a patient’s profile. The few examples discussed here (such as inhibitors of Syk, CaMKIV, and ROCK) have already shown activity in proof-of-principle experiments with immune cells from SLE patients as well as in some animal models. These types of drugs offer additional advantages over the current biologic agents in terms of ease of administration and dosing and will hopefully have fewer side effects than broader immunosuppression. Thus, a gene expression profile could be used to develop an individualized regimen using specific targeted therapies, moving us toward the ultimate goal of personalized medicine for SLE. the rheumatologist
Dr. Lo is a pediatric rheumatology fellow at Children’s Hospital Boston. Dr. Tsokos is chief of the rheumatology division at Beth Israel Deaconess Medical Center and a professor at Harvard Medical School, both in Boston.
References
- Kyttaris VC, Zhang Z, Kampagianni O, Tsokos GC. Calcium signaling in systemic lupus erythematosus T cells: A treatment target. Arthritis Rheum. 2011;63:2058-2066.
- Deng GM, Liu L, Bahjat FR, Pine PR, Tsokos GC. Suppression of skin and kidney disease by inhibition of spleen tyrosine kinase in lupus-prone mice. Arthritis Rheum. 2010; 62:2086-2092.
- Enyedy EJ, Nambiar MP, Liossis SN, Dennis G, Kammer GM, Tsokos GC. Fc epsilon receptor type I gamma chain replaces the deficient T cell receptor zeta chain in T cells of patients with systemic lupus erythematosus. Arthritis Rheum. 2001; 44:1114-1121.
- Tenbrock K, Tsokos GC. Transcriptional regulation of IL-2 in SLE T cells. Int Rev Immunol. 2004; 23:333-345.
- Ichinose K, Juang YT, Crispin JC, Kis-Toth K, Tsokos GC. Suppression of autoimmunity and organ pathology in lupus-prone mice upon inhibition of calcium/calmodulin-dependent protein kinase IV. Arthritis Rheum. 2011; 63:523-529.
- Stohl W, Elliott JE, Li L, Podack ER, Lynch DH, Jacob CO. Impaired nonrestricted cytolytic activity in systemic lupus erythematosus: Involvement of a pathway independent of Fas, tumor necrosis factor, and extracellular ATP that is associated with little detectable perforin. Arthritis Rheum. 1997; 40:1130-1137.
- Lee JH, Wang LC, Lin YT, Yang YH, Lin DT, Chiang BL. Inverse correlation between CD4+ regulatory T cell population and autoantibody levels in paediatric patients with systemic lupus erythematosus. Immunology. 2006;117:280-286.
- Scalapino KJ, Daikh DI. Suppression of glomerulonephritis in NZB/NZW lupus-prone mice by adoptive transfer of ex vivo expanded regulatory T cells. PLoS One. 2009;4:e6031.
- Desai-Mehta A, Lu L, Ramsey-Goldman R, Datta SK. Hyperexpression of CD40L by B and T cells in human lupus and its role in pathogenic autoantibody production. J Clin Invest. 1996; 97:2063-2073.
- Sidiropoulos PI, Boumpas DT. Lessons learned from anti-CD40L treatment in systemic lupus erythematosus patients. Lupus. 2004;13:391-397.
- Wang A, Guilpain P, Chong BF, et al. Dysregulated expression of CXCR4/CXCL12 in subsets of patients with systemic lupus erythematosus. Arthritis Rheum. 2010;62:3436-3446.
- Li Y, Harada T, Juang YT, et al. Phosphorylated ERM is responsible for increased T cell polarization, adhesion, and migration in patients with systemic lupus erythematosus. J Immunol. 2007;178:1938-1947.
- Chong BF, Mohan C. Targeting the CXCR4/CXCL12 axis in systemic lupus erythematosus. Expert Opin Ther Targets. 2009;13:1147-1153.
- Crispin JC, Keenan BT, Finnell MD, et al. Expression of CD44 variant isoforms CD44v3 and CD44v6 is increased on T cells from patients with systemic lupus erythematosus and is correlated with disease activity. Arthritis Rheum. 2010; 62:1431-1437.
- Biswas PS, Gupta S, Chang E, et al. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J Clin Invest. 2010; 120:3280-3295.
- Apostolidis SA, Crispin JC, Tsokos GC. IL-17-producing T cells in lupus nephritis. Lupus. 2011; 20:120-124.
- Anand A, Dean GS, Quereshi K, Isenberg DA, Lydyard PM. Characterization of CD3+ CD4- CD8- T cells in patients with systemic lupus erythematosus: activation markers. Lupus. 2002;11:493-500.
- Mok CC. Biomarkers for lupus nephritis. J Biomed Biotechnol. 2010; 2010:638413. Epub Apr 19.
- Fernandez DR, Telarico T, Bonilla E, et al. Activation of mammalian target of rapamycin controls the loss of TCRzeta in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation. J Immunol. 2009; 182:2063-2073.
- Fernandez DR, Perl A. mTOR signaling: A central pathway to pathogenesis in systemic lupus erythematosus? Discov Med. 2010; 9:173-178.
- Juang YT, Peoples C, Kafri R, et al. A systemic lupus erythematosus gene expression array in disease diagnosis and classification: a preliminary report. Lupus. 2011; 20:243-249.
- Jeffries M, Dozmorov M, Tang Y, Merrill JT, Wren JD, Sawalha AH. Genome-wide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythematosus. Epigenetics. 2011;6:epub ahead of print.
- Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus. Arthritis Rheum. 1990;33:1665-1673.
- Sunahori K, Juang YT, Kyttaris VC, Tsokos GC. Promoter hypomethylation results in increased expression of protein phosphatase 2A in T cells from patients with systemic lupus erythematosus. J Immunol. 2011;186:4508-4517.
- Mishra N, Reilly CM, Brown DR, Ruia P, Gilkeson GS. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J Clin Invest. 2003; 111:539-552.