
In 1998, my friends Gary Firestein, MD, chief of rheumatology and immunology at the University of California, San Diego School of Medicine, and Wim van den Berg, PhD, head of the rheumatology research lab and professor of experimental rheumatology at Nijmegen Centre for Molecular Life Sciences in The Netherlands, and I edited a book called “T Cells in Arthritis.”1 In the introduction, we wrote that “The goal of this book is to outline the major arguments and data suggesting that T cells may, or may not, be central players in the pathogenesis of chronic [rheumatoid arthritis (RA)]. … a great deal is at stake when considering these important questions. Most significant, the direction of future therapeutic interventions depends heavily on how one interprets the pathogenesis of RA.”
Indeed, today T cells are back, and we already act on T-cell interactions in the treatment of rheumatoid arthritis with CTLA4-Ig (Orencia), which inhibits T-cell costimulation by blocking the interactions of CD28 on T cells and B-7 molecules on antigen-presenting cells. This paper will focus on novel cytokine interleukin-17 (IL-17), a central player in immune regulation, and the related Th17 cells. In contrast to the more classical reviews on IL-17, I will provide here a more personal description of how these molecules and concepts have emerged to be so fashionable today.2
The Discovery of IL-17
Once IL-17 arrived on the scene in 1995, its link with RA was immediately established. One of the first reports describing the role of the cytokine IL-17 showed that synoviocytes from RA patients produced IL-6 when exposed to IL-17. This established the first link between IL-17 and inflammation.6
IL-17 had started its life under the name of CTLA-8, a gene product without clear function. At the time of IL-17’s discovery, I was encouraging the Schering-Plough facility near Lyon to use IL-4 to control RA inflammation. The Schering-Plough group was very active in the field of B cells, and the new molecule called CTLA-8 was tested in all the B-cell and T-cell assays in routine use in the laboratory. No effect was observed and the project was going to be dropped until someone suggested testing an effect of RA synoviocytes, which I had available in my laboratory.
This last effort to find a function for IL-17 was successful; induction of IL-6 by synoviocytes was observed, making IL-17 indeed a French discovery. At the same time as I was conducting this work, researchers at Immunex in Seattle (now part of Amgen) reached a similar conclusion from their work on the IL-17 receptor.7 At this early stage of research, inhibitors were obtained—including a mouse anti-human IL-17 monoclonal antibody and an anti-mouse IL-17 soluble receptor also active on human IL-17—so these tools could have been transferred quickly to the clinic as early as 1996.
The link between IL-17 and RA was further extended when we showed that pieces of RA synovium could produce bioactive IL-17. This activity was measured with a bioassay where supernatants from pieces of cultured RA synovium induced a massive production of IL-6 by RA synoviocytes.8 Using the first available monoclonal antibody to block human IL-17, such production was reduced by two-thirds. This robust production of IL-6 using supernatants with IL-17 contrasted with the limited effect of recombinant IL-17 alone.
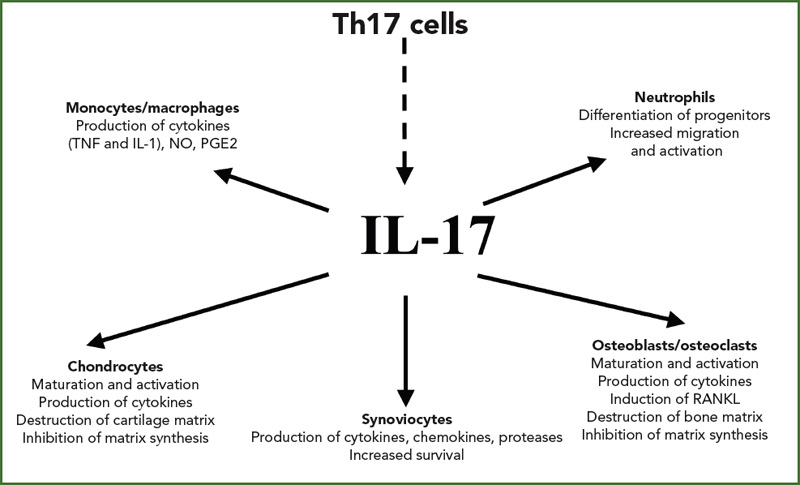
The conclusion of these experiments was that bioactive IL-17, produced by the RA synovium, interacted with other known stimulating cytokines, such as TNF and IL-1, to induce IL-6 production (See Figure 1, right). These interactions were shown to be the consequence of synergy. We know now that IL-17 increases the effects of TNF and IL-1 by increasing the mRNA half-life of genes induced by these cytokines. Observation of synergistic interactions in mesenchymal cells supports the regulatory nature of signals from T cells acting on monocytes and dendritic cells.
The inhibitory effect of IFNγ on IL-17 production and function as clarified in 2003 by Masanori Kawashima, who was working in my laboratory.9 Dr. Kawashima was comparing the effect of IL-12, with or without IL-18, on the expression of IFNγ and IL-17. In the paper published on these experiments, we wrote, “Results for healthy PBMC and RA synovium cells show a discrepancy between IFNγ and IL-17 regulation by IL-12 and IL-18. In particular, IL-12 plus IL-18 synergistically increased the expression of IFNγ mRNA, but decreased that of IL-17, in both PBMC and RA synovium cells.” We concluded that IFNγ had a critical role for inflammation and IL-17 a critical role for destruction.
Dr. van den Berg, among others, has been instrumental in demonstrating the contribution of IL-17 to arthritis in various mouse models. In some models, with dynamic changes in the cell composition of the synovitis, he was able to show that the TNF contribution seen in early disease could be lost in a more chronic phase of the disease, replaced by the key role of IL-17.10 In a paper we published together, he also showed that the addition of IL-4 through gene therapy could inhibit IL-17 production and action and protect against bone destruction by reducing the key interactions between RANK and RANK ligand.11
A New Subset of T cells
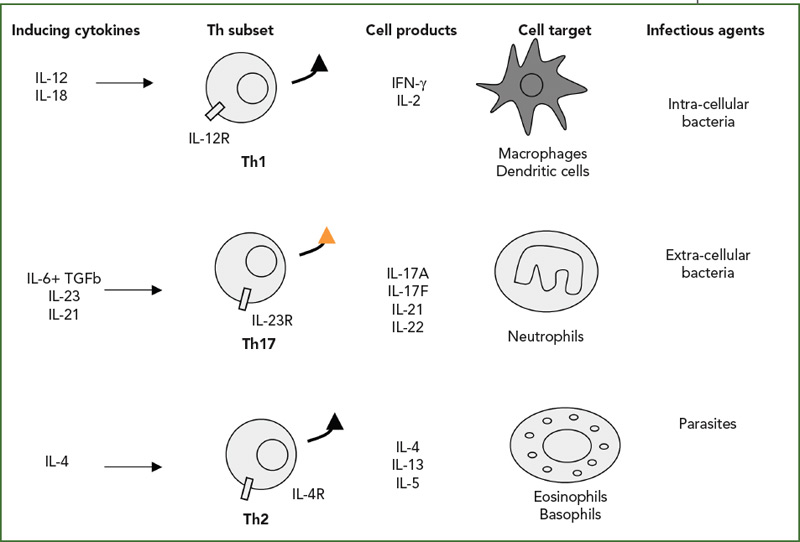
The Th1/Th2 classification was described by my friends Tim Mossman and Bob Kaufman, working then at Dnax in Palo Alto, Calif. Indeed, such simple dichotomy has been useful to classify both mouse and human diseases (See Figure 2, right). According to this classification, infections with intracellular pathogens such as tuberculosis (TB) are Th1 diseases, and asthma and other allergic conditions are Th2 diseases. From the beginning, however, the fixed pattern of T cell function made it difficult to understand chronic diseases. When we looked at a long list of T-cell clones from the RA synovium we had obtained from Jacob Natvig, MD, professor in the Institute of Immunology at the University of Oslo in Norway, it was clear that the IFNγ-producing cells were more common than the Th2 IL-4–producing cells.
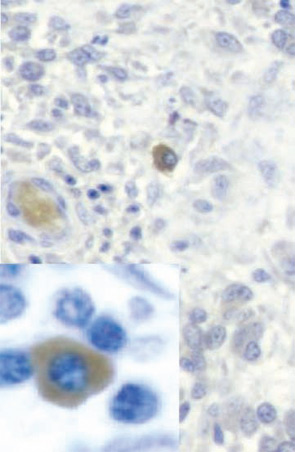
Based on this type of result, RA was classified as a Th1 rather than a Th2 disease. When we added our bioassay for IL-17, clones could be classified more precisely. IL-17–producing cells were never found in IL-4–producing cells but could be found in IFNγ-producing cells. These results, obtained in 1999, are the first description of the production of IL-17 by a different subset of T cells.12 In the RA synovium, IL-17–producing cells have a plasma cell–like morphology (See Figure 3, p. 24).
The official description of the Th17 cells was made in 2005 in the mouse.13,14 This lineage was considered independent from the Th1 lineage. Whereas the key cytokines for the induction of IFNγ are IL-12 and IL-18, the key cytokines for the induction of IL-17 are IL-6 plus TGFβ followed by IL-23. The associated transcription factors are also different, with tBet for Th1 pathway and ROgammat for the Th17 pathway.15
Role of IL-17 in Aspects Still Uncontrolled by TNF Inhibitors
Blocking TNF has been a major step in RA treatment and has produced dramatic improvements in patients not adequately responsive to other DMARDs. In addition to being an important advance in therapy, the efficacy of TNF blockers showed that, although the list of target cytokines is long and forever growing, blocking a single one works, probably by disrupting the synergistic interactions between cytokines. We all know that other interventions are needed to one day cure RA. It is of interest to consider if controlling IL-17 could bring us closer.
The use of TNF inhibitors has been associated with a number of cases of TB, mostly through reactivation. Part of the explanation for the link between TB and TNF inhibition is the presence of a defect in cell-mediated immunity in active RA. Indeed, peripheral blood mononuclear cells from patients with RA respond poorly to the synergistic interactions between IL-12 and IL-18 to produce IFNγ, the key cytokine for protection against intracellular pathogens. Such a defect is related to disease activity as it is partially reversible in responders. IL-17 expression, which is linked to RA disease activity, has an enhancing effect on this immune defect through a specific down-regulation of the IL-12–specific chain of the IL-12 receptor. Lack of a functional receptor leads to reduced response to IL-12 and production of IFNγ, increasing the risk of TB reactivation.
An important limitation of TNF inhibitors is the necessity for continuous therapy. In general, disease recurs a few months after stopping treatment. This indicates that mechanisms upstream or downstream of TNF are still activated and reemerge when TNF inhibition is stopped. IL-17 may have a role in this setting through its effect on synoviocyte survival. Treatment of synoviocytes with IL-17 inhibits activation-induced apoptosis. Furthermore, arthritic mice with chronic streptococcal cell wall arthritis where the IL-17 receptor (IL-17R) has been inactivated have reduced disease activity with increased synoviocyte apoptosis. This is in part related to the enhancing effect of IL-17 on synoviolin expression. Synoviolin is an anti-apoptotic molecule, and elevated levels of synoviolin in RA blood have been associated with lack of response to infliximab.16
Long-lasting remission and even a cure for RA is the next goal of therapy. My friend Vijay Kuchroo, DVM, PhD, professor of neurology at Harvard Medical School in Boston, has established a key point by showing that inflammation itself is able to induce defects in regulatory T cells.17 As indicated above, the classical proinflammatory cytokines IL-6, IL-1, and TNF interact with IL-23 to promote the Th17 pathway. Chronicity follows when IL-17, in turn, inhibits the T regulatory pathway. Inhibition of TNF, which also affects IL-17 production, can partially correct these regulatory defects. It is suggested that control of IL-17, either directly with IL-17–specific inhibitors or upstream by acting on IL-23, may be needed to restore a full regulatory function. This could lead to a reactivation of repair mechanisms, which are completely depressed by inflammation.
Plans for Use of IL-17 Inhibitors
One may ask why it took so much time to consider the use of IL-17 inhibitors in the treatment of RA. In 1995 and 1996, Schering-Plough had a blocking anti–IL-17 antibody and a patent on the protein. Immunex had a soluble IL-17 receptor and a patent for the IL-17 receptor. Since neither company had the full rights to the development of IL-17 inhibitors for RA, it is likely they were unwilling to plan a clinical trial, possibly afraid of a lawsuit from the other competitor. It took 20 years to administer a biological inhibitor of IL-17 to humans. Strangely, this was performed in 2007 by another company, Novartis, without longstanding interests in IL-17.
Before, the choice for intervening in the IL-17 pathway appeared easier, with one ligand, IL-17, and one receptor, IL-17R, to target. Today, the options have increased with the other members of the IL-17 family, which include IL-17A to IL-17F. The IL-17 receptor family has also expanded, with IL-17RA and IL-17RC being critical for the action of IL-17A and IL-17F.
As of today, a demonstration of the beneficial effect of IL-17 inhibition is yet to be obtained in the clinic. Limitations for targeting as a therapy IL-17 could be the low bioactivity of IL-17—at least when used alone—the reduced numbers of IL-17–secreting cells in RA synovium, and the overlap with the effects of TNF. Thus, the position of IL-17 inhibition in the hierarchy of RA therapy is still unclear. In addition, planning RA clinical trials is often affected by tradition. For agencies today, the goal is to be equal or superior to TNF inhibitors. If this practice is followed, then patients resistant to TNF inhibitors will have to be enrolled in initial trials of an anti–IL-17 inhibition. In contrast, trials would need a different design if control of chronicity and induction of repair are the primary endpoints.
Conclusion
The story from the inside shows that basic research can inspire disease-related research, that the development of new therapeutics can be slow and complicated when patent issues are involved, and that the pursuit of fashionable research may not always be productive. This story is also about friendship—it’s been 20 years since Wim, Gary, and I edited our textbook together. Science can be a way to enjoy life, and many of my best research ideas have come from times when I’ve shared a beer with Wim and Gary or, from the comfort of a sandy beach, I’ve watched Gary surf. I look forward to many more years of science and friendship with my colleagues and I wonder how, 20 years from now, we will look at the renewed interest in cells whose importance was once seriously doubted.
Dr. Miossec is professor of immunology and rheumatology at the Hôpital Edouard Herriot in Lyon, France.
References
- Miossec P, van den Berg WB, Firestein GS. T Cells in Arthritis. Boston: Birkhauser; 1998.
- Miossec P. Interleukin-17 in fashion, at last: ten years after its description, its cellular source has been identified. Arthritis Rheum. 2007;56:2111-2115.
- Firestein GS, Zvaifler NJ. Peripheral blood and synovial fluid monocyte activation in inflammatory arthritis. II. Low levels of synovial fluid and synovial tissue interferon suggest that gamma-interferon is not the primary macrophage activating factor. Arthritis Rheum. 1987;30:864-871.
- Firestein GS, Zvaifler NJ. How important are T cells in chronic rheumatoid synovitis?: II. T cell-independent mechanisms from beginning to end. Arthritis Rheum. 2002;46:298-308.
- Miossec P, Naviliat M, Dupuy d’Angeac A, Sany J, Banchereau J. Low levels of interleukin-4 and high levels of transforming growth factor beta in rheumatoid synovitis. Arthritis Rheum. 1990;33:1180-1187.
- Fossiez F, Djossou O, Chomarat P, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593-2603.
- Yao Z, Painter SL, Fanslow WC, et al. Human IL-17: A novel cytokine derived from T cells. J Immunol. 1995;155:5483-5486.
- Chabaud M, Durand JM, Buchs N, et al. Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42:963-970.
- Kawashima M, Miossec P. Heterogeneity of response of rheumatoid synovium cell subsets to interleukin-18 in relation to differential interleukin-18 receptor expression. Arthritis Rheum. 2003;48:631-637.
- Koenders MI, Lubberts E, van de Loo FA, et al. Interleukin-17 acts independently of TNF-alpha under arthritic conditions. J Immunol. 2006;176:6262-6269.
- Lubberts E, Joosten LA, Chabaud M, et al. IL-4 gene therapy for collagen arthritis suppresses synovial IL-17 and osteoprotegerin ligand and prevents bone erosion. J Clin Invest. 2000;105:1697-1710.
- Aarvak T, Chabaud M, Miossec P, Natvig JB. IL-17 is produced by some proinflammatory Th1/Th0 cells but not by Th2 cells. J Immunol. 1999;162:1246-1251.
- Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123-1132.
- Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133-1141.
- Bettelli E, Korn T, Kuchroo VK. Th17: The third member of the effector T cell trilogy. Curr Opin Immunol. 2007;19:652-657.
- Toh ML, Marotte H, Blond JL, et al. Overexpression of synoviolin in peripheral blood and synoviocytes from rheumatoid arthritis patients and continued elevation in nonresponders to infliximab treatment. Arthritis Rheum. 2006;54:2109-2118.
- Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235-238.
