Rheumatology is a discipline that has more than its share of syndromes that are mysterious in both origin and presentation. Inflammation of uncertain origin is one of the conditions that particularly intrigues us. Indeed, we are excited and challenged when patients seek our help when they present with a baffling combination of symptoms such as joint pains, skin rashes of varying description, and fever accompanied by an acute phase response. Some of these conditions have found a name—Behçet’s, Still’s disease, and Schnitzler’s syndrome, for instance—but progress towards identifying their causes, and, of greater importance, finding an effective treatment, has been slow. With progress in immunology, however, some of the mystery is dissipating, and new treatments are on the horizon.
The pioneering studies of Daniel Kastner, MD, PhD, at the National Institutes of Health and others have gained recognition for a group of diseases that are now called the autoinflammatory syndromes; some of theses disease are hereditary and are now much better understood in terms of molecular pathology.1 A common feature of this group of diseases is systemic inflammation in the absence of known infectious agents, often presenting a chronic course that responds only partially to treatment. Some authors also call this process sterile inflammation to distinguish it from sepsis.
A major breakthrough in our understanding of these inflammatory syndromes came with the characterization of the inflammasome. Despite initial skepticism about the nature and role of this subcellular structure, the inflammasome is now widely accepted as a key regulator of inflammatory processes. Importantly, the functional activity of the inflammasome provides an explanation for hitherto difficult to explain syndromes. It is increasingly clear that the inflammasome may be involved in multiple inflammatory processes. The inflammasome also brings back into the limelight the role in disease of interleukin (IL)-1b, a cytokine that has up to now been overshadowed by tumor necrosis factor–α, at least in rheumatology.

Some History
The term “inflammasome” was coined by Jürg Tschopp and colleagues at the University of Lausanne in Switzerland to describe a caspase-activating complex that processes pro–IL-1b (35kd) to mature IL-1b (17kd form). Two research groups, including Tschopp’s, published around the same time that an intracellular complex composed of caspase-1, apoptosis-associated speck-like protein with a caspase-recruitment domain (ASC), and nucleotide binding domain and leucine-rich repeat containing protein (NLRP) is required for IL-1b processing in macrophages.2,3 This initial observation has led to an explosion of studies that have extended our concepts of the inflammasome. As now recognized, the inflammasome can be composed of different pro-inflammatory caspases as well as different NLRP-family proteins, and its substrates include IL-18 and possibly IL-33, cytokines that belong to the IL-1 family.4
Despite the complex names and the different permutations of their component molecules, inflammasome structures share the common property of caspase activation on assembly. The NLRP component is thought to act as a sensor, either of exogenous or endogenous danger signals that are generated by infection and cell stress, to initiate the assembly of the complex, eventually leading to dimerization and activation of the caspases. The NLRPs are part of the NLR (NOD-like receptor) family of proteins that has many members and shares a similar domain structure. Most NLRs are composed of a leucine-rich repeat (LRR) domain, an oligomerization domain, and an effector region that mediates interactions with effector caspases through homotypic interactions that were first identified in apoptosis related caspases (see Figure 1, p. 19). Three families have been identified based on sequence homology and are named the NLRP (or NALP), IPAF, and NOD families (see Figure 2, p. 19).
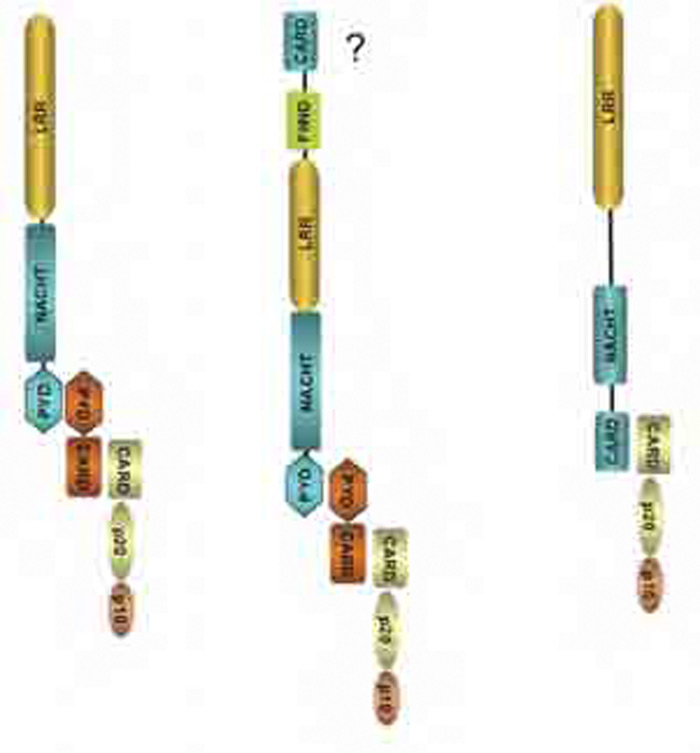
The NLRP1 inflammasome has a CARD domain at its C-terminus and may be able to associate directly with caspases. The NLRP3 inflammasome is composed of ASC and caspase-1, and the IPAF inflammasome does not contain ASC.
How sensing is performed by NLRPs is still poorly understood, but the LRR domain resembles that found in toll-like receptors, which have been shown to have a sensor function for ligands such as lipopolysaccharide. To date, three inflammasomes have been identified: NLRP1, NLRP3, and IPAF. ASC, a component of the NLRP1 and NLRP3 inflammasomes, acts as an adaptor to link the sensor to the effector caspase and is essential for its function. Absence of ASC blocks IL-1b and IL-18 processing. The final component is the pro-inflammatory caspase, which in man could either be caspase-1, -4, -5, or -12. Caspase-1 was previously also known as IL-1 converting enzyme, and its importance in processing IL-1b was described nearly 20 years ago.5,6 The properties of the other caspases have been studied in less detail. These components of the inflammasome are found in the cytoplasm, but there is variation in their tissue distribution, particularly of the NLRs. Immunohistochemistry showed that leucocytes express high levels of NLRP1 and 3, and epithelial cells differentially express these two proteins depending on the tissue.7
The most interesting part of the inflammasome story concerns the signals that lead to its activation. As previously mentioned, the NLR is thought to act as the sensor of endogenous or exogenous danger signals. This sensor function has been conserved throughout evolution, as the plant homologs of NLR, the resistance (R) genes, are critical for the plants’ defense against bacteria, fungi, and viruses. In humans, the list of inflammasome activators is ever expanding and includes components of infectious agents (e.g., Leishmania, Yersinia, Staphylococcus), particles (e.g., microcrystals), metabolic alterations (such as hyperglycemia and adenosine triphosphate concentration), and chemicals. In terms of human health and disease, inflammasome activation plays a key role in cryopyrin-associated periodic syndrome (CAPS), acute gouty inflammation, and the mechanism of action of vaccine adjuvants.
The macromolecular complex (inflammasome) consists of NLRP3, ASC, and procaspase-1. Assembly leads to activation of caspase-1 that in turn cleaves pro–IL-1b to produce biologically active IL-1b. The suggested scheme is based on available data for monosodium urate (MSU)–induced activation of macrophages. Cell binding or uptake of the activator leads to the generation of reactive oxygen species through activation of nicotinamide adenine dinucleotide phosphate hydrogen oxidases. This event activates the NLRP3 inflammasome. MSU crystals may also induce the secretion of ATP that in turn activates P2X7R. On activation of the P2X7 receptor, there is rapid exit of intracellular potassium that triggers the NLRP3 inflammasome. A rise in intracellular calcium is also required for the secretion of processed IL-1b.
Interest in the inflammasome was ignited by the identification of mutations in the NLRP3 gene as the molecular basis of CAPS. Patients with this condition had defied diagnosis for more than two decades until the identification of the genetic mutation and subsequent studies linking dysregulated IL-1b production by constitutive activity of the NLRP3 inflammasome.8-9 As IL-1 inhibitors (e.g., anakinra or IL-1Ra) were readily available, a proof-of-concept study could be rapidly performed, with the striking results we now know.10 These elegant translational studies closed the loop of pathogenesis and established the first link between inflammasome activation and disease in humans and rekindled our interest in the role of IL-1 in human disease.
The next major discovery in this field involved crystal-induced disease with the demonstration that monosodium urate as well as calcium pyrophosphate crystals can activate macrophages to produce IL-1b via the NLRP3 inflammasome.11 The link with gouty inflammation was obvious. Again, a proof-of-concept study using IL-1Ra as inhibitor showed remarkable effects in acute gout,12 the second disease to be linked to inflammasome activation. The third and most recent disease to demonstrate a role for Inflammasome-mediated IL-1b release is diabetes mellitus, where, in fact, a clinical study showing the therapeutic effect of IL-1 inhibition preceded the biochemical studies demonstrating the role of the NLRP3 inflammasome.13-14 In mice that are deficient for components of the NLRP3 inflammasome, the diabetic phenotype is attenuated.
The expanding list of inflammasome activators now include silica and asbestos fibers, vaccine adjuvants such as alum, cells undergoing necrosis, and the contact sensitizer trinitrochlorobenzene (TNCB) (see Table 1, p. 18).15-20 These findings will require confirmatory studies in the human disease setting but suggest that the inflammasome may be involved in myriad disease-associated inflammatory processes.
Important unresolved questions for now relate to what promotes the inflammasome to assemble and how it senses danger signals. Although the NLR component possesses a sensor domain in its LRR region, there is no proof that it makes direct contact with the activating agent. Indeed, there is evidence for a multitude of intermediate cellular changes that link cellular contact with the activator and assembly of the inflammasome complex. Current theories for this process invoke a role for cell stress and the generation of reaction oxygen species, the redox protein thioredoxin binding protein, as well as potassium flux and extracellular ATP acting via the P2X7 receptor.14,21 Identification of the pathway(s) and their cell specificity will be of enormous interest, because they may give further insights into potential targets for drug development as well as disease mechanisms.
A Future for Anti–IL-1 Therapy?
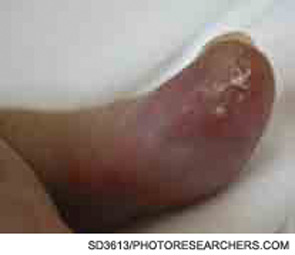
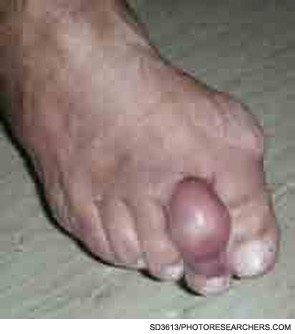
The description of the inflammasome and its role in IL-1b processing has coincided with an expanding application of IL-1 blockade in disease. The discovery of NLRP3 mutations in CAPS patients made IL-1 an obvious target for therapeutic intervention, and the concept has been validated by studies using different IL-1 inhibitors. The results of blocking IL-1 were spectacular in a disease that had hitherto been extremely difficult to manage and had considerable morbidity as well as mortality (see Figures 3 and 4, below right). To date, three molecules have been tested in clinic: IL-1Ra or anakinra, IL-1 Trap or rilonacept, and the anti–IL-1b monoclonal antibody canakinumab. All three agents are highly effective in treating the spectrum of CAPS manifestations in pilot studies as well as in controlled studies, and the treatments were well tolerated.10,22,23
Currently, controlled trials in gout with rilonacept and canankinumab are in progress, and positive results have been reported as abstracts.24,25 It is obviously inappropriate to treat all cases acute gout in this way, but these agents promise to be an alternative therapy in patients who cannot tolerate nonsteroidal antiinflammatory drugs or colchicine. Other conditions have been successfully treated with anakinra, suggesting that inflammasome-mediated IL-1 production has a role in pathophysiology. These conditions include Still’s disease (both juvenile onset and adult types), Schnitzler’s syndrome (a condition of uriticaria, fever, bone pain, arthritis, and an immunoglobin M gammopathy), and acute arthritis due to calcium pyrophosphate dihydrate (CPPD). In all these cases, conventional treatment with antiinflammatories, steroids, and immunosuppressive drugs had not provided long-lasting relief. The successful results with IL-1Ra, sometimes used over prolonged periods, have been invaluable for patients with these conditions. Other indications for IL-1 blockade that may come into clinical reality are based on data from experimental models or early-phase clinical studies; these include diabetes mellitus, the prevention of asbestosis, and in the treatment of certain skin conditions. The concerns about therapy targeting IL-1, apart from efficacy and costs, are the risk for infections and long-term immunosuppression. However, the current data from clinical studies with IL-1 inhibitors have not signaled any major safety concern. The future of anti-IL-1–based treatments looks bright.
In the next few years, we can expect that the field of inflammasome research will continue to expand. Importantly, future studies will determine how these sensors of danger signals may be subverted to maintain chronic inflammation. As inflammation is a process common to many different diseases, from atherosclerosis to Parkinson’s disease, it is obviously important to understand how this process is maintained and how its modulation may change disease course.
Dr. So is professor of rheumatology and Dr. Pazár is in the service of rheumatology, both at the University Hospital Lausanne in Switzerland.
References
- Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: The molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol. 2009;27:621-668.
- Martinon F, Burns K, Tschopp J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417-426.
- Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem. 2002;277:21119-21122.
- Martinon F, Mayor A, Tschopp J. The inflammasomes: Guardians of the body. Annu Rev Immunol. 2009;27:229-265.
- Kostura MJ, Tocci MJ, Limjuco G, et al. Identification of a monocyte specific pre-interleukin 1 beta convertase activity. Proc Natl Acad Sci U S A. 1989; 86:5227-5231.
- Thornberry NA, Bull HG, Calaycay JR, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768-774.
- Kummer JA, Broekhuizen R, Everett H, et al. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem. 2007;55:443-452.
- Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29:301-305.
- Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1 beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319-325.
- Hawkins PN, Lachmann HJ, Aganna E, McDermott MF. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 2004;50:607-612.
- Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237-241.
- So A, Desmedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007;9:R28.
- Larsen CM, Faulenbach M, Vaag A, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517-1526.
- Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136-140.
- Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674-677.
- Li H, Willingham SB, Ting JP, Re F. Cutting edge: Inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J Immunol. 2008;181:17-21.
- Kool M, Petrilli V, De Smedt T, et al. Cutting edge: Alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol. 2008;181: 3755-3759.
- Sharp FA, Ruane D, Claass B, et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci U S A. 2009;106:870-875.
- Iyer SS, Pulskens, WP, Sadler JJ, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A. 2009;106:20388-20393.
- Watanabe H, Gaide O, Petrilli V, et al. Activation of the IL-1 beta-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol. 2007;127:1956-1963.
- Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007; 14:1583-1589.
- Goldbach-Mansky R, Dailey NJ, Canna SW, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355:581-592.
- Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. 2009;360:2416-2425.
- So A, De Meulemeester M, Pikhlak A, et al. Canakinumab (ACZ885) vs. triamcinolone acetonide for treatment of acute flares and prevention of recurrent flares in gouty arthritis patients refractory to or contraindicated to NSAIDs and/or colchicine. Presented at the 75th Annual Scientific Meeting of the ACR; October 2009; Philadelphia.
- Terkeltaub R, Sundy J, Schumacher HR, et al. The IL-1 inhibitor rilonacept in treatment of chronic gouty arthritis: Results of a placebo-controlled, cross-over pilot study. Ann Rheum Dis. 2009;68:1613-1617.
