Editor’s Note: The nomenclature for Wegener’s recently changed from Wegener’s granulomatosis to granulomatosis with polyangiitis (Wegener’s). Beginning with the June 2011 publication of The Rheumatologist, the new nomenclature will be used. See “Medical Societies Ask, What’s in a Name?“ on p. 48 for more on the evolution of disease nomenclature.
Wegener’s granulomatosis (WG) is per definitionem a granulomatous disorder involving the respiratory tract and is usually associated with vasculitis, affecting small to medium-sized vessels and the production of antibodies to neutrophil cytoplasmic antigens (ANCA) directed to the antigen proteinase 3 (PR3).1 Although vasculitis may be its classical feature, WG also may occur as a persistent granulomatous inflammatory process without apparent vasculitis.2 While there is no clear definition of granulomatous disease in WG, this pathological process relates to the formation of masses (often round in appearance on radiograph) or the histological picture of granuloma that is frequently detected within these masses. We know about the vasculitic side of WG’s coin; much less is known about the granulomatous side of WG, however. In this article, we will focus on recent clinical and immunopathological findings attributed to the so-far enigmatic granulomatous process and the probable link to the development of PR3-ANCA and vasculitis.3
Categorizing Disease Stages
WG is commonly considered to be part of the spectrum of disorders characterized by vasculitic lesions throughout the body without a known cause but associated with autoantibodies against PR3-ANCA.4 Therefore, WG is included into the group of diseases known as primary systemic vasculitides and grouped together with others (e.g., microscopic polyangiitis, Churg-Strauss syndrome) also strongly associated with ANCA and “pauci-immune” glomerulonephritis in situ.2 There is strong evidence for the pathogenic role of antibodies against myeloperoxidase (MPO-ANCA) in inducing vasculitis, and this mechanism may also play a role in PR3-ANCA associated vasculitis such as WG.5 However, the clinical and pathological hallmark of WG is not only vasculitis but the coexistence of vasculitis and granuloma, both of which differentiate WG from microscopic polyangiitis. The presence of two distinct pathological processes needs to be considered in categorizing disease stages in WG.
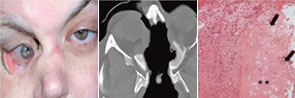
right panel, Arthritis Rheum. 2008;58:834.
Reprinted with permission.
Currently, there are two approaches to categorize disease stages in ANCA-associated vasculitis (AAV). In the United States, it is common to differentiate a limited form of WG in which patients suffer from non–life-threatening vasculitis manifestations from a severe form with life-threatening vasculitic manifestations, such as glomerulonephritis.6 In contrast, the European approach separates a disease stage with granulomatous manifestations of the upper and lower respiratory tract (and with no clinical signs of vasculitis) from systemic disease stages with granulomatous and vasculitic manifestations (early systemic, non–life-threatening; generalized, organ-threatening; and severe with organ failure).7,8
To simplify this categorization, we can identify two forms of WG: 1) a complete form (systemic or generalized WG) that is characterized by both granulomatous and vasculitic manifestations and presents ultimately as the organ- and life-threatening pulmonary–renal vasculitis syndrome; and 2) an incomplete form of WG that is categorized as localized WG. In this form, the disease occurs solely as granulomatous inflammation that is restricted to the upper and/or lower respiratory tract without any other systemic involvement or constitutional symptoms. These two forms differ from previous approaches to clinical classification—which, in turn, has an impact on ideas related to disease pathogenesis and specifically related to the interplay of granulomas inflammation and frank vasculitis.
The previous concept for understanding WG, both clinically and mechanistically, related to chronology. In this scheme, localized WG could appear initially as a short-term and “mild” disease stage (localized WG) before patients increased their level of disease severity with ensuing vasculitic symptoms to develop complete WG. Yet, rarely, localized WG may persist for decades as primarily a chronic granulomatous inflammatory process restricted to the respiratory tract. This aspect of disease has been described only recently in a systematic approach by two European cohort studies.9,10 Furthermore, this necrotizing granulomatous inflammation can cause more severe local and long-term organ damage than previously thought.11
Clinical Spectrum of WG
In the past, the major fatal outcome of untreated and generalized WG was associated with manifestations of vasculitis such as alveolar hemorrhage (due to capillaritis of the lungs) or rapidly progressive glomerulonephritis (due to pauci-immune crescentic glomerulonephritis), leading to vital organ failure within days or weeks. This fatal outcome of full-blown (generalized or severe) WG is less frequent today due to earlier diagnosis and improved treatment strategies. However, the mortality rate is still high (around 11% [range, 2.2%–25%] depending on disease stage and intensity of treatment), especially within the first year of diagnosis.12
Because of improved diagnostic techniques, the growing awareness of orphan diseases in the medical community and the major improvement of therapeutic strategies, WG is now recognized in a wider clinical spectrum that encompasses not only systemic vasculitis manifestations but also chronic granulomatous manifestations of the respiratory tract. Indeed, in the everyday care situation of patients with WG, the majority of patients with “grumbling” and/or relapsing disease suffer from the ongoing granulomatous process rather than from vasculitis.
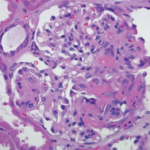
Regarding localized disease manifestation, mucosal ulceration leading to bloody rhinitis is seen by nasal endoscopy during the beginning of the disease; later, septum infiltration and perforation may occur as well as the destruction of the conchae. Granulomatous masses may be detected by MRI imaging in the sinuses and can be accompanied by bone destruction of the adjacent sinus, ethmoidal cysts, and orbita, leading to proptosis and compression of the optic nerve.13 In addition, per vias naturales the masses can obstruct the lacrimal duct or the subglottic region and, less frequently, bronchi, which may cause subsequent poststenotic pneumonia. Via the ethmoidal cysts, the process rarely invades the intracranial space and brain. In the lung, the masses usually appear as round nodules with a tendency to cavitate (see Figure 1, above).
Damage Induced by Granulomatous Inflammation
Although advances in the treatment of WG have resulted in a dramatic reduction in disease-related mortality, patients still experience relapses due to the vasculitis and the granulomatous process and side effects from treatment toxicity.11 Using damage scores to assess the total burden of damage experienced by the patient, it can be demonstrated that, besides the damage associated with malignancy, tissue ischemia, and organ failure, items of damage obviously associated with the granulomatous process were frequently scored and rated as medium to severe damage items.11 Examples of damage related to granulomas include proptosis, pseudotumor, nasal bridge collapse, subglottic/ large airway obstruction, orbital wall destruction, diplopia, and optic nerve atrophy. These findings show that the so-called granuloma include space-occupying lesions and tissue-destructive events.
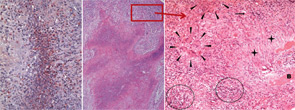
right panel, Rheumatology. 2008;47:1111-1113.
Reprinted with permission.
Localized Granulomatosis as a Persistent Variant of WG
Localized WG may occur not only as a short-term disease stage before generalized disease develops. Recently, this form of disease also has been identified by two European centers as a persistent variant.9,10 In our study, persistent localized WG was observed in 5% of patients in a cohort of 1,024 patients with biopsy-compatible WG followed from 1989 to 2009.9 Only around 50% of the patients were ANCA positive. Localized disease manifestations were characterized by destructive and space-occupying lesions and associated with a high rate of organ damage. Furthermore, nearly all patients with granulomatous involvement (89% over the whole course of follow-up) required medium or highly potent immunosuppression such as methotrexate or cyclophosphamide for remission induction. Cotrimoxazole, which was used in half of the patients as initial treatment, was shown to be insufficient to control disease activity in 72%. In spite of close follow-up and step-up of immunosuppression in cases of persistent or refractory disease activity, 66% of patients acquired at least some kind of organ damage (i.e., saddle nose deformity, septal perforation, bony destruction of sinus and orbital walls).
The findings of this study were confirmed by a smaller cohort from France that included 16 patients with persistent localized disease.10 Persistent localized disease was found to occur at a similar frequency (3.2%). Furthermore, highly potent immunosuppression with cyclophosphamide was required at a similar rate as in the larger study to control localized destructive disease of the ear-nose-throat (ENT) region or mass formation in the lungs.
In summary, a small percentage of WG remains persistently localized to the ENT region (and to adjacent structures), presents with granulomatous-type manifestations within the respiratory tract, which may lead to destruction of bone and cartilaginous structures of the nose (e.g., saddle nose, loss of concha, septum perforation) to invasion of the orbita (orbital pseudotumors), or cranial base (intracerebral lesion) with organ-threatening complications that require more aggressive immunosuppression than previously expected.
Genetic Background of AAV
Interestingly, the incidence of WG is much lower in Japan compared with European countries.14 It may be speculated that differences in genetic background are responsible for the differences in incidences of the AAV. So far, especially from European cohorts, there is evidence that AAV patients share common genetic risk factors but also have their own unique genetic variations, which confer the differences in phenotype. Whereas the PTPN22*620W allele is associated with both WG and MPA, the HLA-DPB1*0401 allele has only been identified as a risk factor for WG, but not for MPA.15–18 Interestingly, HLA-DPB1*0401 is also associated with another granulomatous disease, chronic beryllium disease. It may therefore be speculated that HLA-DPB1*0401 may confer a risk for the development of granulomatous disease, whereas PTPN22*620 W is typically associated with a positive ANCA status and may therefore convey the risk for a vasculitic phenotype. Currently, a European genome-wide association study of WG and MPA is underway to identify common and unique genetic variations in WG and MPA. In the future, a genetic characterization of Japanese populations will be required to understand the different incidence rates of AAV in Europe and Japan.
Granulomatosis of Wegener’s: Histological Features, Immunopathogenic Mechanisms
Whereas immunopathogenic mechanisms of vasculitis are thought to be well established, the driving force and pathophysiological meaning of granulomatous inflammation in WG are unknown.3 We would like to summarize information about histological features and pathomechanisms within granulomatous lesions, in short, granulomatosis.
The classical histomorphologic triad of WG comprises granuloma (see the right panel of Figure 2, p. 41), geographic necrosis (see the middle panel of Figure 2, p. 41), and vasculitis.19–21 Granuloma is defined as a focus of histiocytic cells, chiefly epitheloid cells, with or without multinucleated giant cells and T lymphocytes. In WG, however, the granuloma itself is situated within a dense heterogeneous inflammatory infiltrate of bystander cells (see the right panel of Figure 2, p. 41). In fact, the typically ill-defined WG granuloma is accompanied by numerous neutrophils (PMN), diffusely dispersed macrophages/histiocytes, fibroblasts, plasma cells, and lymphocytes. T and B lymphocytes may be diffusely distributed or organized as follicle-like structures.3,19–21
PMN, aggregating to neutrophilic microabscesses (see the left panel of Figure 2, p. 41)—frequently in the center of epitheloid cell granulomas—undergo cell death, eventually forming areas of geographic necrosis.20 This cell death may occur via NETosis, a cell death program that could play a central role in the induction of autoimmunity.22
Within the inflammatory lesions of granulomatous WG, lymphocytes together with follicular dendritic cells form germinal center-like structures (see the right panel of Figure 2, p. 41), a process very similar to rheumatoid synovium. These follicular structures provide a microenvironment for the development of autoreactive B cells (see below), which may link both hallmarks of WG, the granulomatous lesion as a site of autoantibody production and the autoimmune vasculitis. Surprisingly, another pathogenic process takes place in the granulomatous lesion (i.e., cartilage) (see the right panel of Figure 1, p. 41) and bone destruction (see the right panel of Figure 2, p. 41). This process has marked similarity to events in the rheumatoid synovium.
Autoantigens and Autoantibodies in the Granulomatous Lesion
Autoantibodies to constituents of PMN (e.g., PR3 [PR3-ANCA] or myeloperoxidase [MPO] [MPO-ANCA]) are the serological hallmarks of systemic vasculitis, and PR3-ANCA is strongly associated with WG. PR3 and MPO are abundant in neutrophil extracellular traps (NETs) formed by PMN that undergo nuclear disintegration and decondensation. NETs are web-like structures composed of chromatin bound to positively charged molecules (histones, neutrophil serine proteases). NETs play an important role physiologically and act in antimicrobial defense carried out by PMN.23 NETs have been found in glomerulonephritis of WG both in situ and in the bloodstream.24 Excess formation of NETs and their presentation of a modified PR3 also suggest a way by which dying PMN present in microabscesses and contribute to the necrotic areas in the granulomatous lesion and a subsequent rise of PR3-ANCA.20,25 In addition, a NETs-mediated release of factors such as B-lymphocyte stimulator by PMN could, for instance, promote the activation of B cells.26

We have reported that germinal center-like structures containing T and B cells are often found within granulomatous lesions and hypothesized that germinal center-like structures might be an important element of a microenvironment that supports autoantibody-producing plasma cells (see references 21, 27, and 28 for a review of this). Indeed, we showed an accumulation of heavy chain variable region immunglobulin gene mutations in the endonasal mucosa of WG, pointing towards an influence of local autoantigens, such as PR3 on the generation of autoimmunity.29
An accumulation of replacement mutations can be observed in the complementarity determining 3 (CDR3) region of immunoglobulin genes, with these mutations serving as an indicator of selection and affinity maturation in germinal center-like structures of granulomatous lesions. Distinct offsprings of B lymphocyte clones in nasal mucosa indicated clonal expansion.29,30 Following laser-microdissection of single CD20+ tissue B cells, pairs of variable immunoglobulin light- and heavy-chain genes were isolated, sequenced, and cloned into corresponding vectors. Subsequently, these clones were recombinantly expressed using a baculovirus/insect cell system. Complete immunoglobulins were then tested for their antigen specificity. These studies identified autoantibodies directed against a transmembrane protein and a tetraspanin. Summarizing our initial experiments, we observed that the granulomatous lesion with its germinal center-like structures contains autoantibody-producing cells.30,31 Preliminary studies, detecting a frequent ANCA idiotype (5/7 Id), indicate the presence of PR3-ANCA-producing cell within the granulomatous lesion (unpublished data).
Previously, we demonstrated a predominance of T cells with a skewed Th1-type response in granulomatous lesions of localized WG, which was less prominent in generalized WG (see Figure 3, p. 43).28,32 Interestingly, in vitro co-cultures of PR3-exposed dendritic cells with autologous CD4+ T cells from patients with WG yielded a significantly higher production of interferon-gamma, which may be considered as the initiation of an adaptive immune response through PR3.33 Granulomatous lesions from generalized WG exhibited a Th2-type cytokine profile (see Figure 3, p. 43).34 Furthermore, it is assumed that Th17 cells participate in the pathogenesis of WG; it remains to be determined, however, if Th17 cells play a role in the granulomatous inflammation of WG.35 The current challenge to understand pathogenesis is to develop integrated models for the process of chronic inflammation that consider numerous feedback loops and regulatory networks in which T cell-derived cytokines operate in the granulomatous tissue.
Destruction Takes Place in the Granulomatous Lesion
Whereas damage of nasal cartilage and bone (see Figures 1 and 2, p. 41) is a well-known clinical feature of WG, the pathway of destruction is poorly understood. Some theories have postulated an ischemic etiology due to vasculitis; however, there has been no histopathological proof for mechanism. Moreover, despite cartilage or bone destruction, the majority of nasal granulomatous lesions in WG do not show a vasculitis.
To elucidate this important element of disease, tissue destruction in WG has been explored in a transfer model using immunodeficient pfp/rag2 -/- mice. In studies analogous to experimental models developed for rheumatoid arthritis, nasal biopsies of active WG were co-implanted together with healthy human cartilage. In contrast to controls, implants from nasal mucosa of patients with WG showed massive cartilage destruction mediated by human fibroblasts. It is assumed that matrix metalloproteases released by infiltrating fibroblasts are crucial for destruction. Interestingly, the destruction could be inhibited by corticosteroid treatment of the mice. To summarize the results of these experiments, this is the first model to indicate that inflamed nasal mucosa of patients with WG, resembling the cellular composition of the inflammatory infiltrate has destructive potential; and to provide evidence that destruction occurring in the respiratory tract is induced by a granulomatous cell infiltrate, not vasculitis. The model also implies that fibroblasts are the crucial cell type within granulomatous lesions that may be responsible for destruction.36
Conclusion
Summarizing the results from clinical studies, today we know that the granulomatous inflammation is associated with severe clinical complications. The complications include space-occupying masses leading to proptosis and blindness; destruction of surrounding tissue, cartilage and bone; and obstruction of the trachea (subglottic stenosis) and bronchi. A genetic predisposition may confer the formation of granulomatous lesions in WG. Summarizing the data from experimental (in vitro and in vivo) studies, granulomatous lesions may support the following pathogenic mechanisms: they may create a microenvironment for the induction of cartilage and bone destruction and the development of autoimmunity, eventually leading to the production of PR3-ANCA and to vasculitis. Despite major progress in disease treatment, important questions on pathogenesis remain: Why do granulomatous masses and lesions develop in WG? Additionally, why are there are patients who have only localized WG? Hopefully, future research will provide answers to these fundamental questions and lead to further improvement in the treatment of this serious and vexing disease.
Acknowledgment
We thank E. Csernok, K. Holl-Ulrich, F. Moosig, and P. Lamprecht for their critical discussion.
The authors are part of the Vasculitis Center, Department of Rheumatology and Clinical Immunology, University Hospital Schleswig-Holstein, Campus Lübeck and Klinikum Bad Bramstedt, Germany.
References
- Leavitt RY, Fauci AS, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum. 1990;33:1101-1107.
- Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37:187-192.
- Bacon PA. The spectrum of Wegener’s granulomatosis and disease relapse. N Engl J Med. 2005;352:330-332.
- Reinhold-Keller E, Beuge N, Latza U, et al. An interdisciplinary approach to the care of patients with Wegener’s granulomatosis: Long-term outcome in 155 patients. Arthritis Rheum. 2000;43:1021-1032.
- Xiao H, Heeringa P, Hu P, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidas cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110:955-963.
- Stone JH; Wegener’s granulomatosis etanercept trial research group. Limited versus severe Wegener’s granulomatosis: Baseline data on patients in the Wegener’s granulomatosis etanercept trial. Arthritis Rheum. 2003;48:2299-2309.
- Rasmussen N, Jayne DR, Abramowicz D, et al. European therapeutic trials in ANCA-associated systemic vasculitis: Disease scoring, consensus regimens and proposed clinical trials. Clin Exp Immunol. 1995;101:S1:29-43.
- Hellmich B, Flossman O, Gross WL, et al. EULAR recommendations for conducting clinical studies and/or clinical trials in systemic vasculitis: Focus on anti-neutrophil cytoplasm antibody-associated vasculitis. Ann Rheum Dis. 2007;66:605-617.
- Holle JU, Gross WL, Holl-Ulrich K, et al. Prospective long-term follow-up of patients with localised Wegener’s granulomatosis: Does it occur as persistent disease stage? Ann Rheum Dis. 2010;69:1934-1939.
- Pagnoux C, Stubbe M, Lifermann F, et al. Wegener’s granulomatosis strictly and persistently localized to one organ is rare: Assessment of 16 patients from the French vasculitis study group database. J Rheumatol. 2011;38:475-478.
- Seo P, Min YI, Holbrook JT, et al. Damage caused by Wegener’s granulomatosis and its treatment: Prospective data from the Wegener’s granulomatosis Etanercept Trial (WGET). Arthritis Rheum. 2005;52:2168-2178.
- Little MA, Nightingale P, Verburgh CA, et al. Early mortality in systemic vasculitis: Relative contribution of adverse events and active vasculitis. Ann Rheum Dis. 2010;69:1036-1043.
- Muhle C, Reinhold-Keller E, Richter C, et al. MRI of the nasal cavity, the paranasal sinuses and orbits in Wegener’s granulomatosis. Eur Radiol. 1997;7:566-570.
- Fujimoto S, Uezono S, Hisanga S, et al. Incidence of ANCA-associated primary renal vasculitis in the Miyazaki Prefecture: The first population-based, retrospective, epidemiologic survey in Japan. Clin Am Soc Nephrol. 2006;1:1016-1022.
- Jagiello P, Aries P, Arning L, et al. The PTPN22 620W allele is a risk factor for Wegener’s granulomatosis. Arthritis Rheum. 2005;12:4039-4043.
- Carr EJ, Niederer HA, Williams J, et al. Confirmation of the genetic association of CTLA4 and PTPN22 with ANCA-associated vasculitis. BMC Med Genet. 2009;10:121.
- Heckmann M, Holle JU, Arning L, et al. The Wegener’s granulomatosis quantative trait locus on chromosome 6p21.3 as characterized by tagSNP genotyping. Ann Rheum Dis. 2008;67:972-979.
- Arning L, Holle JU, Harper L, et al. Are there specific genetic risk factors for the different forms of ANCA-associated vasculitis? Ann Rheum Dis. 2011;70:707-708.
- Lie JT. Wegener’s granulomatosis: Histological documentation of common and uncommon manifestations in 216 patients. Vasa. 1997;26:261-270.
- Holl-Ulrich K. Histopathology of systemic vasculitis. Pathologe. 2010;31:67-76.
- Mueller A, Holl-Ulrich K, Lamprecht P, et al. Germinal centre-like structures in Wegener’s granuloma: The morphological basis for autoimmunity? Rheumatology. 2008;47:1111-1113.
- Remijsen Q, Berghe TV, Wirawan E, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011;21:290-304.
- Papayannopoulos V, Zychlinsky A. NETs: A new strategy for using old weapons. Trends Immunol. 2009;30:513-521.
- Kessenbrock K, Krumbholz M, Schönermarck U, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15:623-625.
- Urban C, Ermert D, Schmid M, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009;5:e1000639.
- Scapini P, Bazzoni F, Cassatella M. Regulation of B-cell-activating factor (BAFF)/B lymphocyte stimulator (BLys) expression in human neutrophils. Immun Lett. 2008;116:1-6.
- Mueller A, Voswinkel J, Hallof A, et al. Immunophenotypic characterization of germinal centre-like structures in Wegener’s granulomatosis. APMIS. 117:126 [abstract].
- Aries P, Lamprecht P, Gross WL. Wegener’s granulomatosis: A view from the granulomatous side of the disease. IMAJ. 2005;7:768-773.
- Voswinkel J, Mueller A, Kraemer JA, et al. B lymphocyte maturation in Wegener’s granulomatosus: A comparative analysis of VH genes from endonasal lesions. Ann Rheum Dis. 2006;65:859-864.
- Voswinkel J, Assmann G, Held G, et al. Single cell analysis of B lymphocytes from Wegener’s granulomatosis: B cell receptors display affinity maturation within the granulomatous lesions. Clin Exp Immunol. 2008;154:339-345.
- Thurner L, Müller A, Cerutti M, et al. Wegener’s granuloma harbors B lymphocytes with specificities against a proinflammatory membrane protein and a tetraspanin. J Autoimmun. 2011;36:87-90.
- Müller A, Trabandt A, Gloeckner-Hofmann K, et al. Localized Wegener’s granulomatosis: Predominance of CD26 and IFN-gamma expression. J Pathol. 2000;192:113-120.
- Csernok E, Ai M, Gross WL, et al. Wegener autoantigen induces maturation of dendritic cells and licences them for Th1 priming via the protease-activated receptor-2 pathway. Blood. 2006;107:4440-4448.
- Balding CEJ, Howie AJ, Drake-Lee AB, et al. Th2 dominance in nasal mucosa in patients with Wegener’s granulomatosis. Clin Exp Immunol. 2001;125:332-339.
- Abdulahad WH, Lamprecht P, Kallenberg CGM. T-helper-cells as new players in ANCA-associated vasculitides. Arthritis Res Ther. Submitted.
- Kesel N, Laudien M, Holl-Ulrich K, et al. Xenografted nasal mucosa from Wegener’s granulomatosis patients induces destruction of implanted human cartilage in immunodeficient mice. Arthritis Rheum. 2010;62(10 Suppl):S283 [abstract].
- Aries PM, Both M. Images in clinical medicine. Destructive eye lesions in Wegener’s granulomatosis. New Engl J Med. 2005; 352:392.
- Holl-Ulrich K, Both M, Gottschlich S, Gross WL, Aries PM, Lamprecht P. Clinical images: Saddle nose deformity caused by destructive granulomatous inflammation in Wegener’s granulomatosis. Arthritis Rheum. 2008;58:834.