Aleksandar Mijatovic / shutterstock.com
ATLANTA—At the ACR/ARP 2019 Annual Meeting, several widely renowned experts across an array of specialty subjects provided a comprehensive and compelling review of advances in the understanding, diagnosis and treatment of a number of rheumatologic conditions.
Sjögren’s Syndrome
Frederick Vivino, MD, FACR, chief of rheumatology at Penn Presbyterian Medical Center and professor of clinical medicine at the University of Pennsylvania, Philadelphia, discussed important topics related to Sjögren’s syndrome.
Dr. Vivino noted that, in 2016, the ACR and the European League Against Rheumatism (EULAR) developed a set of classification criteria for Sjögren’s syndrome.1 Although these classification criteria are not intended as a direct means of diagnosing the condition, but are meant to serve as criteria for enrollment in clinical trials, they do serve to inform the clinician of what to look for and evaluate in patients suspected of having primary Sjögren’s syndrome.
The criteria are based on the weighted sum of five items: anti-SSA/Ro antibody positivity; focal lymphocytic sialadenitis with a focus score of ≥1 foci/4 mm2; an abnormal ocular staining score of ≥5 (or van Bijsterveld score of ≥4); a Schirmer’s test result of ≤5 mm/5 min; and an unstimulated salivary flow rate of ≤0.1 mL/min.
Dr. Vivino made several important points regarding the use of these criteria, including the importance of having an experienced and meticulous pathologist perform the focus score calculation when evaluating for focal lymphocytic sialadenitis, and that patients with isolated anti-SSB/La antibody positivity should undergo lip biopsy, because these patients are less likely to have Sjögren’s syndrome.
Although clinical, laboratory & imaging signs may point to Behçet’s, the diagnosis is ultimately clinical, without one confirmatory test.
The most potentially mortal complication in Sjögren’s syndrome is the increased risk of lymphoma, and Dr. Vivino noted that approximately 5–7% of patients with Sjögren’s syndrome will develop non-Hodgkin’s B cell lymphoma within 10 years of diagnosis. Thus, rheumatologists must screen for consistent swollen major salivary glands and have a relatively low threshold for performing biopsy in these patients.
Other manifestations of Sjögren’s syndrome, such as dry eyes and dry mouth, frequently rely on symptomatic rather than disease-modifying therapy. It is imperative that patients with dry mouth use sodium fluoride-containing toothpaste to prevent dental caries. With respect to sialogogues, such as pilocarpine and cevimeline, a good general approach is to start at a low dose and slowly titrate upward to avoid side effects, and to instruct patients to take the dose after meals.
For dry eyes, a key distinction is determining if the issue stems from increased evaporation of tears (i.e., Meibomian gland dysfunction) or from decreased production (i.e., tear deficiency), because the treatment varies on the basis of the exact mechanism.
With regard to immunomodulatory or immunosuppressive medications, the evidence supporting efficacy of these treatments has been equivocal. Hydroxychloroquine is commonly used in the treatment of patients with Sjögren’s syndrome; however, a set of treatment guidelines from 2016 noted relatively weak evidence demonstrates this medication is effective in ameliorating fatigue in patients with Sjögren’s syndrome.2
With respect to the treatment of musculoskeletal pain, there is weak evidence for efficacy of leflunomide, sulfasalazine and azathioprine—all of which may be considered after hydroxychloroquine and methotrexate—and rituximab is typically reserved for vasculitis, recurrent or severe parotid gland swelling, recalcitrant arthritis, pulmonary disease and neuropathy.
Advances in Scleroderma
Lorinda Chung, MD, MS, director of the Scleroderma Center and co-director of the Multidisciplinary Rheumatologic Dermatology Clinic at Stanford University School of Medicine, Palo Alto, Calif., spoke about advances in scleroderma.
A key element of monitoring cutaneous disease activity in scleroderma relies on the use of the modified Rodnan Skin Score (mRSS), which is a measure of skin thickness on a scale of 0–3 (higher score representing thicker skin) at 17 body sites, with a possible total score of 0–51. Dr. Chung noted that several risk factors have been identified as predictors of progression of cutaneous disease in scleroderma, and these include low baseline mRSS, presence of synovitis at baseline and anti-RNA polymerase III antibody positivity. However, the mRSS is sometimes insufficient on its own to enable overall disease progression to be tracked in patients with scleroderma and may not be the most comprehensive way to evaluate efficacy of treatments in clinical trials.
Dr. Chung noted that, in 2016, the ACR developed the Composite Response Index in Systemic Sclerosis (CRISS) to better address these needs.3 The CRISS uses a two-step process that begins with the question: Has the patient demonstrated worsening cardiac, pulmonary or renal manifestations of scleroderma?
If the answer is yes, specifically with regard to new scleroderma renal crisis, a decline of 15% or more in forced vital capacity (FVC) in patients with interstitial lung disease, new pulmonary hypertension on right heart catheterization or new left ejection fraction of less than 45%, then the patient is noted not to have improved on whichever treatment they are using.
If the answer is no, then the mRSS, percent-predicted FVC, patient and physician global assessments, and the Health Assessment Questionnaire Disability Index (HAQ-DI) are used to calculated a CRISS score to indicate the likelihood the patient has improved on treatment.
Dr. Chung explained that use of the CRISS score in clinical trials may hold a great deal of potential to better evaluate if treatments are efficacious in patients with scleroderma.
Another important clinical study in recent years was the Scleroderma: Cyclophosphamide or Transplantation (SCOT) trial, in which the authors compared myeloablative CD34+ selected autologous hematopoietic stem-cell transplantation with immunosuppression by means of 12 monthly infusions of cyclophosphamide in patients with scleroderma.4 The primary endpoint in this study was a global rank composite score that included death, event-free (respiratory, renal or cardiac failure) survival, FVC, HAQ-DI and mRSS. In an intention-to-treat analysis, 67% of pairwise comparisons using the global score favored transplantation over cyclophosphamide at 54 months.
It has been noted that the treatment-related mortality in the transplantation group was 6% at 72 months, compared with 0% in the cyclophosphamide group, and that the composite score used in this study has not been widely used in other studies. Dr. Chung explained that some patients with severe cutaneous disease who have not done well on methotrexate, mycophenolate mofetil or cyclophosphamide may be candidates for myeloablative autologous hematopoietic stem cell transplantation.
Despite only brief mention, it is also important to note that nintedanib, a tyrosine kinase inhibitor, is the first medication\ approved by the U.S. Food & Drug Administration for the treatment of interstitial lung disease associated with scleroderma.
RA
Michael Weinblatt, MD, associate director of the Center for Arthritis and Joint Diseases and John R. Riedman Professor of Medicine at Brigham and Women’s Hospital, Boston, provided an overview with regard to rheumatoid arthritis (RA). Dr. Weinblatt discussed how increased body mass index and diabetes can be associated with nonalcoholic steatohepatitis and that this condition can predispose patients on methotrexate to liver function test abnormalities. In patients on this medication who experience such side effects as gastrointestinal symptoms or oral ulcers, Dr. Weinblatt advocated for the use of leucovorin 5 mg weekly (8–24 hours after the weekly dose of methotrexate) to aid in reducing these side effects.
For patients on oral methotrexate who have not found benefit at a dose of up to 20 mg weekly, split dosing—in which half the weekly methotrexate dose is given and then, from 8–24 hours later, the second half of the weekly dose is administered—can be considered.
In addition to these tips from clinical practice, Dr. Weinblatt also discussed how Janus kinase (JAK) inhibitors represent an interesting and important relatively new category of medication to be explored in the treatment of rheumatoid arthritis. In 2017, the Oral Rheumatoid Arthritis Trial (ORAL) demonstrated that tofacitinib and methotrexate combination therapy was non-inferior to adalimumab and methotrexate combination therapy in the treatment of rheumatoid arthritis in patients with an inadequate response to methotrexate. It should be noted, however, that tofacitinib monotherapy was not shown to be non-inferior to either combination.5
Dr. Weinblatt cited evidence that lack of response early in the course of treatment with tofacitinib (i.e., within one to three months of starting therapy) does predict a low probability of achieving low disease activity, thus patients who do not show clinical improvement after being on tofacitinib for 4–12 weeks may be less likely to benefit from longer periods on this treatment.6 Dr. Weinblatt noted that recent safety concerns over possible increased risk of pulmonary embolism and death in patients being treated with tofacitinib will need to be closely examined and, with the recent FDA approval of the JAK inhibitor upadacitinib, exploring this issue is of even greater importance.
MAS
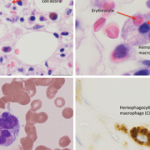
Edward Behrens, MD, Joseph Lee Hollander Chair in Pediatric Rheumatology, and chief, Division of Rheumatology, The Children’s Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, provided a discussion of macrophage activating syndrome (MAS) and hemophagocytic lymphohistiocytosis (HLH).
Dr. Behrens explained that HLH is an aggressive and life-threatening syndrome of excessive immune activation. Primary HLH, also known as familial hemophagocytic lymphohistiocytosis (FHL), is caused by a genetic mutation at either one of the FHL foci or in genes responsible for one of several immunodeficiency syndromes. Secondary HLH is the term used to describe patients without a known familial mutation who develop HLH due to an acute trigger, such as an autoimmune disease, viral illness or lymphoma. MAS indicates the form of HLH that occurs primarily in patients with juvenile idiopathic arthritis or other rheumatic conditions, such as systemic lupus erythematosus; the term reactive hemophagocytic syndrome has also been used in these cases.
Dr. Behrens explained that one challenging issue in this field is how best to diagnose these conditions, especially when comparing primary and secondary forms of HLH. In 2007, the HLH-2004 diagnostic guidelines were introduced and described several criteria that can be used to help establish a diagnosis of HLH.7 However, although these criteria include low/absent NK (natural killer) cell activity, Dr. Behrens stated the function of NK cells is much more relevant in genetic rather than secondary cases of HLH.
In addition, although HLH is one of a few conditions that will result in a serum ferritin level of >10,000 ng/mL, some research indicates that serum heme oxygenase-1 (HO-1) may be of more use in differentiating secondary hemophagocytic syndrome from other similar hematological conditions.8 With respect to activity level in secondary HLH and MAS, work indicates that free interleukin (IL) 18 may be pathogenetic and may serve both as a biomarker for activity and a potential therapeutic target.
Lupus
Susan Manzi, MD, MPH, co-director of the Lupus Center of Excellence and Chair of the Department of Medicine of West Penn Hospital, Allegheny Health System, Pittsburgh, led the discussion of lupus diagnosis and treatment in 2019. This year is of note in the world of lupus because the ACR and EULAR have published recommendations for systemic lupus erythematosus (SLE) classification criteria, and this publication uses a positive anti-nuclear antibody (ANA) in a titer of ≥1:80 as a required entry criterion.
The panel specifically recommends testing by immunofluorescence on human epithelial type 2 (HEp-2) cells or a solid phase ANA screening immunoassay with equivalent performance. By using a combination of clinical and immunologic criteria for which there is not a better explanation than SLE, these classification criteria have demonstrated excellent sensitivity and specificity compared with the sensitivity and specificity of ACR 1997 and Systemic Lupus Erythematosus International Collaborating Clinics (SLICC) 2012 criteria for SLE classification (96.1% and 93.4%, 82.8% and 93.4%, 96.7% and 83.7%, respectively).10
Dr. Manzi also pointed out that one of the most exciting areas of new research in lupus is the Accelerating Medicines Partnership (AMP) program, a collaboration between the National Institutes of Health (NIH), pharmaceutical companies and nonprofit organizations meant to develop new ways of identifying and validating promising biological targets for diagnostics and drug development.
In this vein, a 2017 article described the way single-cell RNA sequencing (scRNA-seq) was used in renal and skin biopsy tissue from patients with lupus.11 One of the key findings in this study was that cumulative expression profiles of single cell keratinocytes from nonlesional, non-sun-exposed skin of patients with lupus nephritis showed upregulation of interferon (IFN) inducible genes compared with keratinocytes isolated from healthy controls, indicating the possibility of using scRNA-seq analysis of skin biopsies as a biomarker of renal disease.
The AMP study groups have undertaken many ongoing studies that seek to further expand knowledge in the fields of lupus and rheumatoid arthritis, and the rheumatology community hopes results from these studies will aid in understanding the pathogenesis of these conditions, help identify surrogate markers for disease activity, and perhaps better classify subpopulations of patients within these broader disease categories.
Finally, on the subject of IFN gene signatures and the role that IFN may play in the pathophysiology of lupus, Dr. Manzi did point out that anifrolumab, a monoclonal antibody against the type I IFN receptor, met the primary endpoint (British Isles Lupus Assessment Group based Composite Lupus Assessment (BICLA) at week 52) in the phase 3 TULIP 2 trial after failing to meet the primary endpoint (SLE Responder Index 4, or SRI4) in the phase 3 TULIP 1 trial.12
Gout
Tuhina Neogi, MD, PhD, Section Chief of Rheumatology at Boston University School of Medicine, Boston, described treatment approaches and areas in need of improvement with regard to the management of gout. Dr. Neogi noted that >50% of patients with gout require a dose of allopurinol >300 mg/day to achieve their goal serum uric acid level.
The goals for treatment are defined as a serum uric acid level of less than 6 mg/dL and, for some patients (i.e. those with tophi), the goal is lower.13 Allopurinol and febuxostat are both options in the initial management of gout, but Dr. Neogi noted the FDA has added a boxed warning to febuxostat indicating increased risk of death compared with allopurinol, with an increased risk of heart-related death and death from all causes with febuxostat.
Dr. Neogi also noted this risk of heart-related and all-cause mortality will need to be further studied, especially because many patients with gout have many co-morbidities, including cardiovascular disease or risk factors for such disease.
Behçet’s
Yusuf Yazici, MD, assistant professor, Department of Medicine (Rheumatology), Hospital for Joint Diseases and Medicine, New York, discussed Behçet’s disease. One of the most important takeaways from this talk was that, although clinical, laboratory and imaging signs may point to Behçet’s, the diagnosis is ultimately clinical, without one confirmatory test. Clinicians must consider mimics of the condition, such as inflammatory bowel disease.
Approximately 98% of patients with Behçet’s have oral ulcers, and these occur primarily on the lips, gingiva, cheeks and tongue, with typical healing within 15 days after onset of each ulcer, according to Dr. Yazici. The genital ulcers in Behçet’s may include scarring scrotal ulcers and most often appear as punched-out lesions.
Cutaneous manifestations of the condition include papulopustular acne—especially in atypical locations in older patients—pathergy, erythema nodosum and superficial thrombophlebitis; when this last entity is present, it is often associated with major vessel involvement with vasculitic disease.
Pulmonary artery aneurysms are the most common arterial complication of Behçet’s disease.
Approximately 5–10% of patients with Behçet’s will have central nervous system involvement, and this can take the form of dural venous sinus thrombosis or parenchymal involvement.
With respect to ocular involvement, anterior uveitis is common, and hypopyon can be seen as well.
IgG4-Related Disease
John Stone, MD, MPH, director of clinical rheumatology in the Rheumatology, Allergy and Immunology Division of Massachusetts General Hospital, Boston, gave the final lecture of the day, on IgG4-related disease (IgG4-RD). Through a series of patient stories, Dr. Stone illustrated how the so-called Mikulicz’s disease, described in 1888 by Dr. Johann von Mikulicz-Radecki in a patient with bilateral, painless and symmetrical swelling of the lacrimal, parotid and submandibular glands may, in fact, have been what we today recognize as IgG4-RD.
IgG4-RD can manifest as inflammation and fibrosis of these glandular structures, as well as exocrine and endocrine pancreatic failure, renal failure, retroperitoneal fibrosis, chronic hepatobiliary failure and even disease that appears like that seen in granulomatosis with polyangiitis.
In sharing a patient story, Dr. Stone described how the condition can be treatment responsive with therapy that most often includes corticosteroids and frequently can include rituximab, but that cumulative organ damage can still occur as described.
The most classic findings on histopathology include lymphoplasmocytic infiltration, storiform fibrosis and obliterative thrombophlebitis, but these features are not always present. Dr. Stone explained that necrotizing vasculitis and primary granulomatous inflammation should not be seen on pathology and serve as exclusion criteria for the diagnosis.
If characteristic clinical and histopathologic findings are present, then the number of IgG4-positive plasma cells/high-powered field can be calculated, and the thresholds for these numbers are tissue specific, he said. There should also be an IgG4+/IgG+ plasma cell ratio of >40%. ACR/EULAR classification criteria for IgG4-RD were published in January.
Conclusion
As clearly shown, the Review Course was wide-ranging, informative, and extremely helpful to clinicians and scientists, and it is likely the knowledge shared will help improve the care of many patients and inspire additional research questions to be explored in the years to come.
Jason Liebowitz, MD, completed his fellowship in rheumatology at Johns Hopkins University, Baltimore, where he also earned his MD. He is now in practice with Arthritis, Rheumatic, & Back Disease Associates, New Jersey.
References
- Shiboski CH, Shiboski SC, Seror R, et al. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjögren’s syndrome: A consensus and data-driven methodology involving three international patient cohorts. Arthritis Rheumatol. 2017 Jan;69(1):35–45.
- Carsons SE, Vivino FB, Parke A, et al. Treatment guidelines for rheumatologic manifestations of Sjögren’s syndrome: Use of biologic agents, management of fatigue, and inflammatory musculoskeletal pain. Arthritis Care Res (Hoboken). 2017 Apr;69(4):517–527.
- Khanna D, Berrocal VJ, Giannini EH, et al. The American College of Rheumatology provisional composite response index for clinical trials in early diffuse cutaneous systemic sclerosis. Arthritis Care Res (Hoboken). 2016 Feb;68(2):167–178.
- Sullivan KM, Goldmuntz EA, Keyes-Elstein L, et al. Myeloablative autologous stem-cell transplantation for severe scleroderma. N Engl J Med. 2018 Jan 4;378(1):35–47.
- Fleischmann R, Mysler E, Hall S, et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): A phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet. 2017 Jul 29;390(10093):457–468.
- van Vollenhoven RF, Lee EB, Fallon L, et al. Tofacitinib in rheumatoid arthritis: Lack of early change in disease activity and the probability of achieving low disease activity at month 6. Arthritis Care Res (Hoboken). 2019 Jan;71(1):71–79.
- Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007 Feb;48(2):124–131.
- Miyazaki T, Kirino Y, Takeno M, et al. Serum HO-1 is useful to make differential diagnosis of secondary hemophagocytic syndrome from other similar hematological conditions. Int J Hematol. 2010 Mar;91(2): 229–237.
- Weiss ES, Girard-Guyonvarc’h C, Holzinger D, et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood. 2018 Mar 29;131(13):1442–1455.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 2019 Sep;71(9):1400–1412.
- Der E, Ranabothu S, Suryawanshi H, et al. Single cell RNA sequencing to dissect the molecular heterogeneity in lupus nephritis. JCI Insight. 2017 May 4;2(9).
- Furie RA, Morand EF, Bruce IN, et al. Type I interferon inhibitory anifrolumab in active systemic lupus erythematosus (TULIP-1): A randomized, controlled, phase 3 trial. The Lancet Rheumatology. 2019;1(4):Pe208–Pe219
- Khanna D, Fitzgerald JD, Khanna PP, et al. 2012 American College of Rheumatology guidelines for management of gout. Part 1: Systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012 Oct;64(10):1431–1446.