The complexity, power, and vastness of these “worlds within worlds” have led to the emerging field of microbiome, which has extended our view of self and transformed the common notion of individuality. Rather than unique and distinct individuals, according to emerging ideas, we should consider ourselves as ecosystems in which multiple species and their genes interact with each other in intricate ways to allow mutual survival.
I could not sufficiently wonder at the intrepidity of these diminutive mortals, who durst venture to mount and walk upon my body […] without trembling at the very sight of so prodigious a creature as I must appear to them.
—Jonathan Swift, describing Gulliver’s reaction to the Lilliputians, Gulliver’s Travels
The term “microbiome,” which was proposed a decade ago by Nobel laureate Joshua Lederberg, identifies the totality of microbes (commensals and pathogenic), their genomes (metagenome), and environmental interactions in a defined community or biological niche. Until recently, the microbial elements of biology have been all but ignored as determinants of health and disease beyond the obvious setting of infection.1
Most recently, and following the same logic, the National Institutes of Health Human Microbiome Project and the European MetaHit consortium have embraced the idea that it is impossible to fully understand human health and disease unless this collective human–microbiome “superorganism” is better studied and defined.2,3 This concept is attractive in understanding the pathogenesis of many rheumatic and autoimmune diseases and, more specifically, in rheumatoid arthritis (RA), which has long been regarded as a complex polygenic entity with an endless search for triggering factors.4
Dwarves Standing on the Shoulders of Giants
RA is a systemic, inflammatory autoimmune disorder. It is currently considered a multifactorial disease, in which multiple genes and environmental factors act in concert to cause pathological events.5 Despite recent advances in understanding molecular pathogenesis and developing more effective treatments, the etiology of RA remains mysterious, and treatment relies on agents that are broad in their action and fail to induce cure. Since the observation in the mid-1980s that the major histocompatibility complex class II alleles (“shared epitope” hypothesis) contribute to RA susceptibility, many other genetic variants associated with disease have been discovered through genome-wide association studies; these variants include PTPN22 and CTLA4, to name only two.4,6 Although genetic studies have improved our understanding of disease susceptibility and provided insight into functional genomics, it has become clear that genetic effects are insufficient to account for differences in the incidence of RA (for instance, RA concordance in monozygotic twins is at best 15%). A gene–environment interaction, including its biological consequences, is therefore a more plausible explanation.
Among environmental risk factors studied in relationship with RA development, the most notable examples include hormones, smoking, alcohol, and infection. Multiple viruses and bacteria have been associated with disease incidence but, to date, causation could not be established. Given the central role of host–microbe interactions, massively parallel DNA-sequence analyses of human microbiomes now offer the potential for important new insights relevant to autoimmune processes. One of the more intriguing environmental covariates modulating autoimmunity involves bidirectional crosstalk between the host and the oral and intestinal microbiomes (see Figure 1).
Figure 1
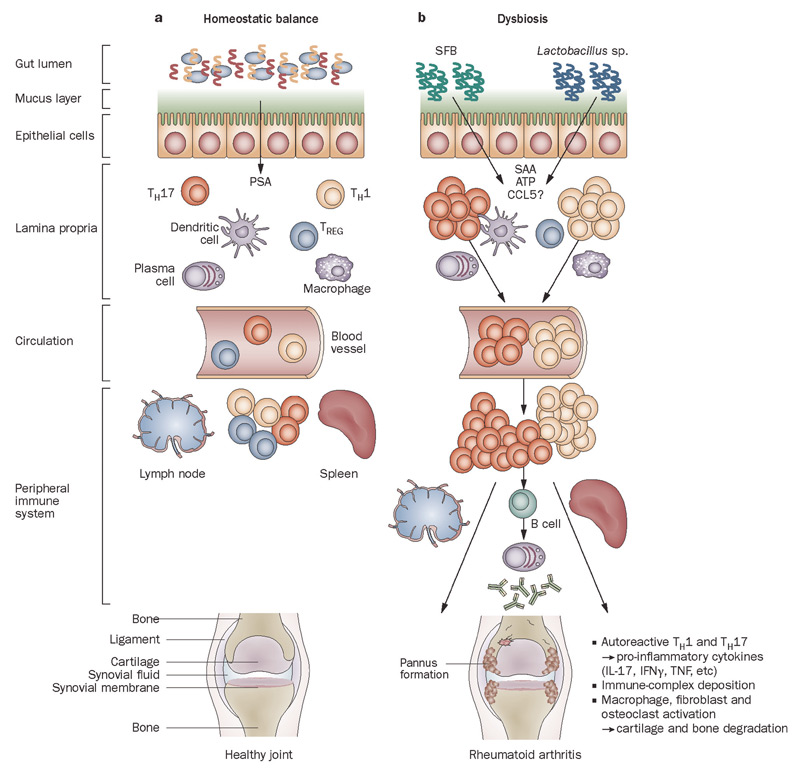
Host–microbiota interactions in health and inflammatory arthritis. A: In healthy individuals, a well balanced host–microbial cross-talk is essential for the maintenance of homeostasis. A thick mucus layer and epithelial cells prevent direct contact with the gut-associated immune cells, which constantly survey the contents of the intestinal lumen and eliminate undesired antigens. Commensal bacteria, such as Bacteroides fragilis, can activate pro tolerogenic machinery. A specific cell wall component, PSA, is sufficient to induce TREG-cell activation, IL-10 production, and TH17-cell repression to avoid uncontrolled inflammation. B: When either genetic or environmental factors alter the balance in the microbiota composition, dysbiosis ensues. Potentially harmful micro-organisms (such as SFB or Lactobacillus) predominate and local expansion of proinflammatory cells (TH17 cells, TH1 cells and others) occurs via different molecules (such as ATP, SAA, or CCL5 signaling). These autoreactive T cells migrate to peripheral immune compartments and activate B cells to differentiate into autoantibody-producing plasma cells. These cells and antibodies then migrate to synovial tissue where the inflammatory cascade is amplified through the activation of effector components, including macrophages, fibroblasts, osteoclasts, cytokines, and proteinases. If self-perpetuating, this process can lead to arthritis and pannus formation.
Abbreviations: ATP, adenosine-5′-triphosphate; CCL5, CC-chemokine ligand 5; IFNγ, interferon γ; IL-17, interleukin-17; PSA, polysaccharide A; SAA, serum amyloid A; SFB, segmented filamentous bacteria; TH1, type 1 T helper cell; TH17, type 17 T helper cell; TREG, regulatory T cell.
Source: Nat Rev Rheumatol. 2011;7:569-578. Reproduced with permission of Nature Pub. Group.
The roots for this appealing notion can be traced many centuries back. In the 1600s, and in the midst of the Great Plague, a devoted and simple Dutch tradesman named Antony van Leeuwenhoek managed to see beyond the tangible. His innovation was a combination of magnifying lenses and metallic skeletons which gave birth to a peculiar creature that would modify science forever: the microscope. With this instrument, van Leeuwenhoek made his most fascinating discovery. Diminutive forms of life, invisible to the bare eye, populated our body cavities and most surfaces on earth.
Struggling to convince the Royal Society, van Leeuwenhoek observed the plaque between his own teeth “with great wonder, that in the said matter there were many very little living animalcules, very prettily a-moving.”
It would take two centuries, however, to prove these animalcules responsible for the origin of infectious processes. With Koch and Pasteur leading and rationalizing the nascent field of microbiology, most medical disciplines then turned into an “infectious rush era.” The logic followed that, since bacteria caused all cardinal signs of inflammation in affected sites, rheumatic diseases were also inevitably a deleterious effect of microorganisms. Theories flourished, and multiple lines of investigation suggested a link between oral or intestinal microbes and RA. Soon after, arthritis deformans was considered to be due to tuberculosis; the focal sepsis hypothesis claimed that RA was caused by periodontal flora; and the toxemic factor idea proposed a role in diseases of substances produced by intestinal bugs.1,7-9 The first half of the 20th century, without the means to prove any of these hypotheses (mainly because one in five bacteria are noncultivable), saw thousands of teeth extractions and even dozens of colectomies performed, hoping to achieve the ultimate cure for RA. Ultimately, none of those expectations materialized, and surgical solutions for RA were gradually abandoned.
Most recently (although debated), paleopathological and epidemiologic evidence has suggested that RA originated in the New World and spread through a transmissible vector to the rest of the globe after 1492.10 These conclusions are based on the lack of skeletal remains with RA-like features outside of America and the high incidence of disease in Amerindians compared with Caucasians, Europeans, Africans, and Asians. Clinical studies implicating specific bacterial agents as triggers for RA have mostly relied on indirect evidence (serological and biased polymerase chain reaction methods) and circumstantial observations.
Who Needs Culture, Anyway?
What theoretically appeared reasonable and necessary to advance the understanding of the microbiome has, nevertheless, encountered challenges. Central to these challenges is the inability to culture and grow approximately 80% of bacteria found in our microbiomes. Whether related to oxygen conditions or nutritional requirements, the immense majority of bacteria cannot survive if they leave their natural habitats. Because this deportation means death for many species, the DNA sequencing technology revolution has enabled the unimaginable: to bypass the need to isolate living organisms for their identification (see Figure 2).
Figure 2
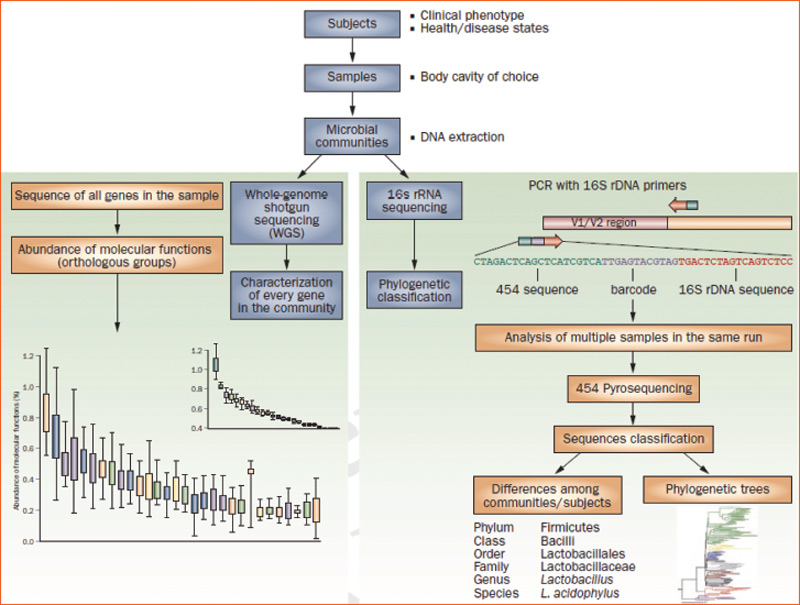
Culture-independent genomic analysis of the human microbiome. Culture-independent techniques have advanced our capacity to survey complex microbial communities in human samples. Well-characterized individuals (healthy and diseased) are asked to donate samples for microbiome analyses. Two metagenomic sequencing approaches are utilized. Conserved and variable 16S rRNA genomic regions are amplified and subjected to pyrosequencing. The resulting sequences are then aligned, filtered, and compared with publicly available databases of 16S rRNA sequences, enabling taxonomic classification of bacteria present or absent in a given sample. Whole genome shotgun sequencing provides information that enables identification of genes present and allows for subsequent comparison of enzymatic pathways and functions represented among different samples. Enzymatic databases are also available to assist in the identification of protein function, enabling the richness and diversity of functional capacities provided by the microbiome to be assessed.
Abbreviations: PCR, polymerase chain reaction; rRNA, ribosomal RNA.
Source: Nat Rev Rheumatol. 2011;7:569-578. Reproduced with permission of Nature Pub. Group.
To assess the microbiome, a highly conserved gene found in all species of bacteria and archaea—the 16S ribosomal RNA (16S rRNA) gene, or 16S—can be targeted, amplified, and utilized for phylogenetic studies.11 Since the gene is specific to bacteria and simultaneously contains hypervariable regions, sequencing 16S permits not only its separation from other prokaryotic and eukaryotic cells within a given sample, but also an unbiased taxonomic tool to differentiate bacterial species among themselves. Next-generation sequencing platforms, such as 454, Illumina, and others, allow for bar-coded, massively parallel 16S rRNA sequencing of millions of sequence determinations in a single experiment. If, a decade ago, the isolation and identification of a given species took months to achieve, dozens of samples containing thousands of different species each can now be sequenced and classified in one day. But this approach only helps with the question of “Who is there?”—in other words, what is the community microbial composition in terms of relative abundance?
To address an even more complex matter (“What are the microbes doing?”), it is now feasible to perform whole-genome shotgun sequencing of a given microbiome (metagenome). Using the same sequencing platforms as in 16S surveys, we also can elucidate the vast array of enzymatic and metabolic pathways generated by these communities, and, with these data, begin to understand their function and behavior. Multiple studies have found that a significant number of intestinal microbiome genes are simply not encoded by the human genome. Among these are genes whose products are essential for the biological development and well-being of humans, including enzymes involved in metabolism of otherwise indigestible polysaccharides, amino acids, and xenobiotics, and the production of proteins associated with the maturation and regulation of the immune system. Concomitantly, an enormous computational biology (and biologist) capacity, along with sophisticated bioinformatic tools and true multidisciplinary efforts are required to filter, align, assemble, and annotate the colossal amount of data and metadata generated and make scientific sense of it.
The Microbiome and the Host Response
More than a thousand bacterial species inhabit our intestinal tract.3 Humans and most mammals in the animal kingdom develop all vital organs and mature under strict sterile conditions, protected by the uterine cavity. Indeed, perturbation by any microorganism often can lead to embryonic malformation or death. Immediately after birth, biologic chaos ensues. Vaginal or skin bacteria (depending on delivery mode) engraft the newborn’s intestines and other body surfaces. Throughout the first year of life, species richness and diversity is rather variable. However, with the introduction of solid foods at the end of this period, the gut microbiota expands, stabilizes, and adopts the complexity of the adult communities.12
To date, more than a thousand different species (and many more strains) can be classified into a dozen divisions. Interestingly, only two bacterial divisions (Bacteroidetes and Firmicutes) dominate the gut microbiome. This raw catalog of animalcules is, nonetheless, insufficient. In order to study associations with health and disease, a critical question needed to be addressed: Is there a core microbiome? Do adult human beings share a set of bacteria that could be considered a “normal” reference? The answer appears remarkable. Humans can be clustered into three groups (or enterotypes) according to their microbiome signature.13 In healthy individuals from industrialized nations, these enterotypes are independent of any (studied) host factors. The basis for this evolutionary pressure remains uncertain. Nevertheless, the symbiotic coexistence between microbiota and the host, along with the factors involved in these cross-communications, are becoming better understood. The central question remains: How are we simultaneously tolerating such an antigenic burden of necessary bacteria while fighting undesired ones?
Recent data support the notion that, in health, the gut microbiota signals through the mucosal-associated lymphoid tissue and shapes immune responses to promote a homeostatic state. Somehow along the evolutionary process, mammals have learned to utilize these pathways to coexist with this huge antigenic load. A physicochemical and immune barrier also separates the two worlds. A thick mucus layer, antimicrobial proteins and secretory immunoglobulin-A, serves as levees in the biological dike to prevent a flood of undesired flora into this system. A tightly adherent epithelial cell column forms a broader dam to further contain bacterial invasion. If still insufficient, the intestinal lamina propria contains the highest concentration of immune cells in mammals. Bacterial proteins and surface molecules such as lipopolysaccharides and other microbe-associated molecular pattern factors interact with toll-like receptors (TLRs) and related pattern recognition receptors in the host immune cells. Dendritic cells, macrophages, and natural killer cells continuously survey the lumen to process, and phagocyte antigens and mount an innate response.
The ultimate balancing act between the host and the microbiome comes from CD4+ T-cells inhabiting the lamina propria. A necessary state of gut mucosal inflammation is driven by T-helper effector cells such as Th1, Th2, and Th17 and their respective signature cytokines, interferon gamma, and interleukins (IL-) 4/13 and 17/22). A tolerogenic subset of T cells, the regulatory T cells (Tregs), actively suppresses and neutralizes potential uncontrolled reactions through IL-10 and transforming growth factor beta. Most recently, it has become clear that a crucial aspect of equilibrium maintenance is directly related to the way the adaptive immune system reacts. Seminal work by Ivanov et al and Round and Mazmanian has recently changed the way we think about the innate and adaptive immunity dichotomy.14,15 Single intestinal commensal bacteria are directly capable of activating CD4+ T-cells and tilting the balance towards pro- or antiinflammatory responses. Segmented filamentous bacteria (SFB), for example, are sufficient to activate Th17 cells, while a specific polysaccharide of Bacteroides fragilis can promote tolerogenic effects via Treg stimulation (see Figure 3).
The Enemy Within?
When the relative phylogenetic composition of the microbiome is altered, a state of dysbiosis can ensue, and the local immune system can shift towards a proinflammatory milieu (see Figure 4). If this reaction is contained and self-regulated, the outcome is typically helpful, and undesirable pathogens can be cleared. When this state of dysbiosis perpetuates, local inflammation results (as in the case of inflammatory bowel disease) and eventually, deleterious systemic effects arise in a predisposed host. Important elements of this model have received support from both classic and recent compelling studies in animal models, particularly those involving germ-free conditions (animals raised in sterile environments and subsequently challenged with different gut bacteria or their antigens). Notable examples of these studies include Kohashi et al and Bjork et al in adjuvant arthritis, work by Taurog et al in HLA-B27 transgenic rats, and reports by van den Broek et al in streptococcal cell wall-induced arthritis rats.16-19
Figure 4
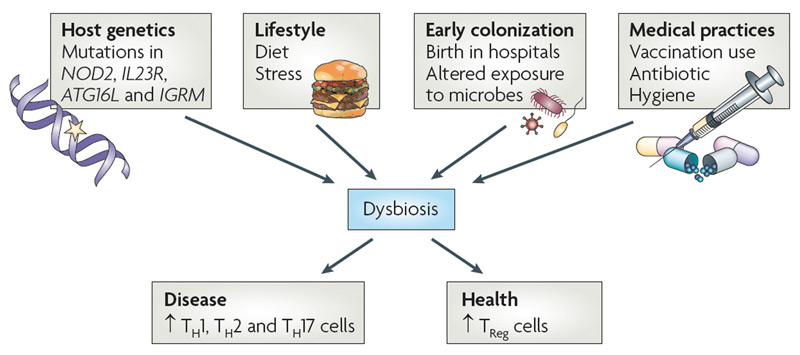
Proposed causes of dysbiosis of the microbiota. The composition of the microbiota can shape a healthy immune response or predispose to disease. Many factors can contribute to dysbiosis, including host genetics, lifestyle, exposure to microorganisms, and medical practices. Host genetics can potentially influence dysbiosis in many ways. An individual with mutations in genes involved in immune regulatory mechanisms or proinflammatory pathways could lead to unrestrained inflammation in the intestine. It is possible that inflammation alone influences the composition of the microbiota, skewing it in favor of pathobionts. Alternatively, a host could “select” or exclude the colonization of particular organisms. This selection can be either active (as would be the case of an organism recognizing a particular receptor on the host) or passive (the host environment is more conducive to fostering the growth of select organisms). Selection of pathobionts by the host could tip the balance in favor of inflammation. Diet and stress also have the potential to influence the microbiota103. Birth in the sterile environment of hospitals can protect from exposure to dangerous pathogens, but can also prevent early exposure to health-promoting bacteria. Overuse of vaccination and antibiotics, which do not distinguish between pathogenic or symbiotic microorganisms, could adversely alter the microbiota.
Abbreviations: ATG16L, autophagy-related gene 16-like; IGRM, immunity-related GTPase family, M; IL23R, interleukin-23 receptor; NOD2, nucleotide-binding oligomerization domain 2; Th, T helper; TReg, regulatory T.
Source: Nat Rev Immunol. 2009;9:313-323. Reproduced with permission of Nature Pub. Group.
The intricate mechanism by which the gut microbiome may modulate systemic disease was recently demonstrated in mouse models of arthritis. When raised under germ-free conditions, both the IL-1 receptor antagonist-knockout (IL1rn-/-) mice and the K/BxN T-cell receptor transgenic model are resistant to inflammatory arthritis.20,21 The introduction of a single gut commensal bacterium (Lactobacillus and SFB, respectively) is sufficient for the development of joint disease. Interestingly, in both models, it is the overactivation of Th17 cells and the dampening of Treg function (via dendritic cells and TLR signaling) that drive systemic autoimmune disease. This model of dysbiosis, leading to local immune imbalance in favor of a proinflammatory Th17-driven state to ultimately promote distal autoimmunity, is not unique to RA. Experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis, is abrogated in germ-free conditions, and also with antibiotic or probiotic treatment.22 Tolerogenic Tregs are responsible for these protective responses. In contrast, EAE susceptibility and severity are enhanced by the administration of bacteria such as Porphyromonas gingivalis and SFB.
Conclusions
The role of microbiome in autoimmunity appears to be far-reaching and important. Clear disease examples of the influence of gut–joint axis include spondyloarthropathies (i.e., reactive arthritis and inflammatory bowel disease–related arthritis), jejunoileal bypass arthritis, and Whipple’s disease.
New sequencing tools and experimental approaches have brought novel insights into the mechanisms behind autoinflammatory processes. Indeed, it is now possible to find and identify thousands of bacteria (and their function), providing an unprecedented level of detail never before witnessed. Animal models of RA have shown the capacity of specific commensal bacteria to activate proinflammatory cells, which in turn initiate and perpetuate deleterious effects in the joint. The clinical implications of these discoveries, along with the idea that humans harbor distinct enterotypes, open a new perspective in rheumatic and autoimmune research. Well-characterized studies utilizing DNA-parallel sequencing, shotgun analyses, and animal models to elucidate possible dysbiosis states in RA and related conditions are in early stages.23
If a distinct microbiota or enterotype is identified, it would then be possible to speculate whether a particular microbiome triggers or drives autoimmunity in genetically predisposed individuals. Identification of “pathogenic” commensal organisms could provide insights into the environmental triggers of diseases such as RA and lead to a new understanding of disease pathogenesis, perhaps leading to novel approaches for thereby. Proving causation, however, will require in-depth mechanistic studies involving “humanization” of mouse models to allow selective colonization with candidate bacteria and study of consequent host immune reaction to balance alteration. Perhaps van Leeuwenhoek’s animalcules were the answer all along.
And maybe, from time to time, the inhabitants of the microbiome, like Gulliver’s diminutive mortals described in Swift’s classic, may not tremble at the sight of such a prodigious creature.
Dr. Scher is clinical instructor of medicine at the New York University School of Medicine and director of the Microbiome Center for Rheumatology and Autoimmunity at NYU Hospital for Joint Diseases in New York. Dr. Abramson is professor of medicine and pathology and director of the division of rheumatology, New York University School of Medicine and NYU Hospital for Joint Diseases.
References
- Lederberg J, McCray A. ‘Ome sweet ‘omics–A genealogical treasury of words. The Scientist. 2001;15:8.
- NIH HMP Working Group, Peterson J, Garges S, Giovanni M, et al. The NIH Human Microbiome Project. Genome Res. 2009;19:2317-2323.
- Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59-65.
- Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205-1213.
- Klareskog L, Catrina AI, Paget S. Rheumatoid arthritis. Lancet. 2009;373:659-672.
- Stahl EA, Raychaudhuri S, Remmers EF, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42:508-514.
- Osler W. The Principles and Practice of Medicine, Designed for the Use of Practitioners and Students of Medicine. 6th ed. New York, London: D. Appleton and Company, 1905.
- Tilley H. Discussion on focal sepsis as a factor in disease. Proc R Soc Med. 1926;19:6-11.
- Warden CC. The toxemic factor in rheumatoid arthritis. Cal State J Med. 1909;7:299-301.
- Rothschild BM., Turner KR, DeLuca MA. Symmetrical erosive peripheral polyarthritis in the Late Archaic Period of Alabama. Science. 1988;241:1498-1501.
- Weisburg WG, Barns SM, Pelletier DA, Lane DJ. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 1991;173:697-703.
- Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol. 2011;9:279-290.
- Arumugam M, Raes J, Pelletier E, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174-180.
- Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485-498.
- Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313-323.
- Kohashi O, Kuwata J, Umehara K, Uemura F, Takahashi T, Ozawa A. Susceptibility to adjuvant-induced arthritis among germfree, specific-pathogen-free, and conventional rats. Infect Immun. 1979;26:791-794.
- Bjork J, Kleinau S, Midtvedt T, Klareskog L, Smedegard G. Role of the bowel flora for development of immunity to hsp 65 and arthritis in three experimental models. Scand J Immunol. 1994;40:648-652.
- Taurog JD, Richardson JA, Croft JT, et al. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med. 1994;180:2359-2364.
- Van den Broek MF, van Bruggen MC, Koopman JP, Hazenberg MP, van den Berg WB. Gut flora induces and maintains resistance against streptococcal cell wall-induced arthritis in F344 rats. Clin Exp Immunol. 1992;88:313-317.
- Abdollahi-Roodsaz S, Joosten LA, Koenders MI, et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest. 2008;118:205-216.
- Wu HJ, Ivanov I, Darce J, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32:815-827.
- Ochoa-Reparaz J, Mielcarz DW, Begum-Haque S, Kasper LH. Gut, bugs, and brain: Role of commensal bacteria in the control of central nervous system disease. Ann Neurol. 2011;69:240-247.
- Scher JU, Ubeda C, Pillinger M, et al. Characteristic oral and intestinal microbiota in rheumatoid arthritis (RA): A trigger for autoimmunity? [abstract #1390] Arthritis Rheum. 2010;62(Suppl):S577-S578.