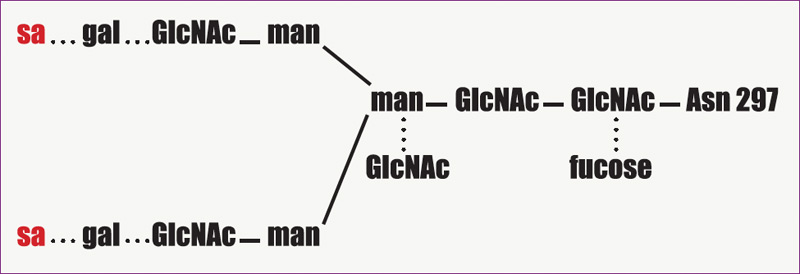
An additional way that IVIg may modulate inflammation is the accelerated degradation of pathogenic IgG by saturation of the neonatal Fc receptor (FcRn), so named because it was initially identified in the neonatal intestinal epithelium. This IgG receptor aids in the transfer of IgG from mother to fetus and also protects IgG from degradation. FcRn is known to exist in many adult tissues, including intestinal epithelium, muscle, and skin. In states of hypergammaglobulinemia, (e.g., following high-dose IVIg), the FcRn is presumably saturated and the catabolism rate of the pathogenic antibody is accelerated. In murine models of autoimmune-mediated skin blistering diseases (e.g., bullous pemphigoid) administration of high-dose IVIg drastically reduces pathogenic IgG levels and prevents blistering. In contrast, mice deficient in FcRn are resistant to the development of blistering in this model and administration of high-dose IVIg does not confer any additional protective effect.9