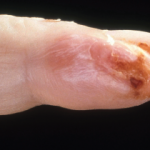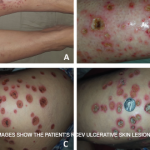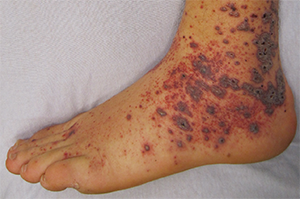
Petechia/purpura on the low limb due to medication induced vasculitis.
James Heilman, MD/wikipedia.com
WASHINGTON, D.C.—The vast majority of the attention given to vasculitis at the ACR/ARHP Annual Meeting, year after year, focuses on ANCA-associated vasculitis and large-vessel vasculitis, said Philip Seo, MD, MHS, director of the Johns Hopkins Vasculitis Center and moderator of the 2016 ACR Review Course titled, Neglected Vasculitis. That leaves out a lot.
“These are important diagnoses,” Dr. Seo said. “There have been a lot of advances made over the last several years. But does that mean there are lot of forms of vasculitis out there that you are going to run into on a regular basis that don’t get any love from the ACR? It’s hard to have an opinion about diagnoses that you don’t see on a daily basis.”