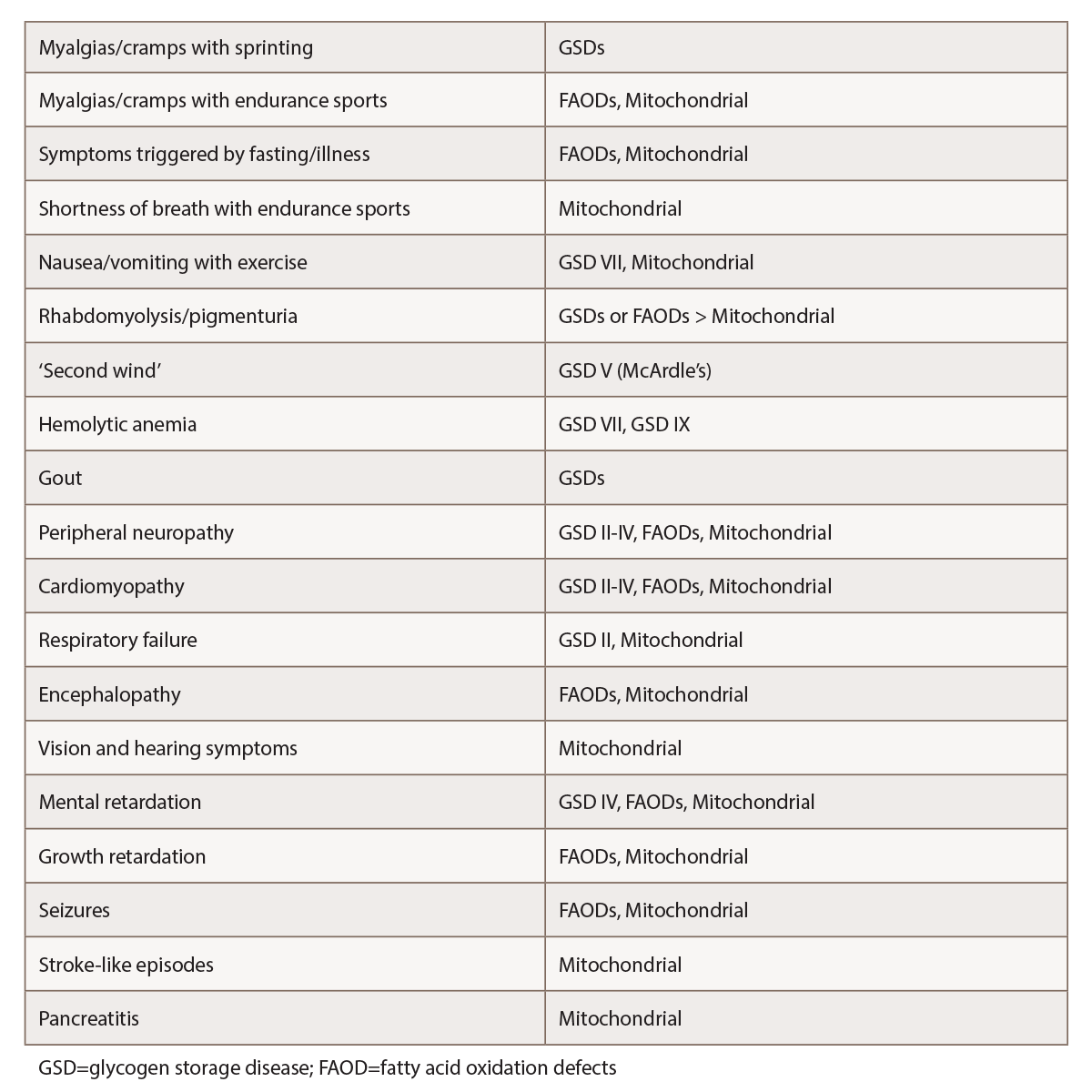
(click for larger image) Table 1: Selected Features Suggestive of Metabolic Myopathies1,2,4,5
(may have myopathic symptoms only)
The physical
The physical also plays a foundational diagnostic role for these patients. “This includes a proper neurological examination with mental status, cranial nerves, motor, reflexes, sensory and gait,” says Dr. Tarnopolsky. A fixed, motor weakness may be present in cases of McArdle’s disease or mitochondrial disorders. In contrast, muscular dystrophy will typically present with obvious muscle weakness, although sometimes the only finding is exercise-induced cramps without other obvious clinical clues.6
Dr. Tarnopolsky explains that for people with fat oxidation defects and glycogen storage diseases, the physical exam is often completely normal (though if a patient has just experienced rhabdomyolysis, they may initially be achy and weak). In contrast, patients with muscular dystrophy or inflammatory myopathy are more likely to display symptoms like shoulder girdle weakness, winging of the scapula, difficulty abducting the arms and difficulty with hip flexion.
The joint and skin exam are also key in establishing or ruling out a potential inflammatory myopathy. “In the metabolic myopathies and the structural myopathies, you are not going to have your Gottron’s rash, your heliotrope rash, your shawl sign, your holster sign, the splinter hemorrhages—all the other things that usually accompany the inflammatory myopathies,” says Dr. Tarnopolsky.
Assessing for muscle atrophy and muscle hypertrophy is also helpful. “Becker and some of the limb girdle muscular dystrophies have enlarged calf muscles, and they are
usually quite strikingly enlarged; whereas, dysferlin and ANO5 type Miyoshi/limb girdle myopathies usually have very thin calf muscles,” says Dr. Tarnopolsky. He notes that muscle bulk changes, such as these, are much more likely to indicate a genetic structural myopathy than an inflammatory or a metabolic myopathy.
Some patients with mitochondrial disease will also have a fixed myopathy. “To help identify those cases, usually there are cranial nerve findings, such as optic atrophy, sensorineural hearing loss, ptosis and ophthalmoparesis, none of which are features of inflammatory myopathy.”
Initial laboratory clues
Guided by clues from the history and physical, initial basic lab work may include tests for CK, myoglobin, basic renal and liver function, lactate, urate, vitamin D, thyroid function, complete metabolic panel (CMP), aldolase, anti-nuclear antibodies, fasting acylcarnitine profile and urine organic acids, as well as tests for inflammation and infection.3,5,7 Dr. Finsterer notes that reviewing all previous tests can be challenging, but is an essential part of the diagnostic process.
Dr. Tarnopolsky recommends the CK as an inexpensive and simple test that can begin to sort out a fixed genetic structural myopathy from an inflammatory, metabolic or other kind of myopathy. “Usually, with glycogen storage disease and [FAOD] patients, the CK is going to go up with metabolic stress, and it usually comes back to normal,” explains Dr. Tarnopolsky. In contrast, CK findings from someone with a structural myopathy such as Becker or limb girdle will show a consistently elevated CK. He says, “If you follow the CK, it stays pretty similarly elevated; so if it’s 2,000 one time, it’s 2,300 the next time.” An inflammatory myopathy, if left untreated, will often show a CK that continues to go up over time.
One exception to this general pattern is the McArdle’s disease patient. “Unlike many metabolic myopathies, where the CK goes up with stress and comes back down to normal, the McArdle’s patient’s CK often stays persistently elevated. And that’s why it can look more like an inflammatory disease or muscular dystrophy,” Dr. Tarnopolsky explains. These patients may also be unusual in that they may not present with the classic exercise-induced symptoms and may appear to be chronically weak, with only minimal activity-induced symptoms.
Dr. Tarnopolsky says a blood acylcarnitine test can be particularly helpful if someone has symptoms induced by fasting or exercise and a FAOD is suspected. As he notes, “A normal acylcarnitine provides pretty good evidence that you have ruled out a fatty oxidation defect.”
He also notes that a fasting lactate can be helpful if a rheumatologist notes anything suspicious on the neurological exam, because an elevated lactate may indicate a mitochondrial myopathy. “If for example, you saw optic atrophy, ptosis, ophthalmoparesis on your exam and you are thinking mitochondrial disease, we would also add plasma amino acid testing. About 20% of [mitochondrial] patients will have an elevated alanine.” He says a metabolic screen on the urine, including metabolic acids, is also indicated in such a situation, because there is often an increase in 3-methylglutaconic acid.
If an inflammatory myositis is still in the differential, Dr. Tarnopolsky suggests a myositis panel. “These are reasonably sensitive and specific. If you had a positive Jo-1 antibody or one of the other classic markers for inflammatory myopathy, it would be very unlikely for that to occur in a metabolic myopathy or structural myopathy.”
Second-line investigations
Follow-up second-line studies depend on previous findings, clinical preference and institutional availability of certain tests. Historically, the next move would typically be investigation via electromyography (EMG) or nerve conduction studies, both of which we would expect to be essentially normal for glycogen storage disease, fat oxidation disorders and most mitochondrial disorders. EMG is often abnormal in muscular dystrophy and inflammatory myopathies. A muscle biopsy is often the next potential test, providing important diagnostic clues regarding potential metabolic myopathies, genetic structural myopathies or inflammatory myopathies.
However, some have suggested moving genetic testing to an earlier point in the process, at least in some instances. For example, some have argued that next-generation sequencing via a rhabdomyolysis gene panel may be more appropriate than traditional muscle biopsy and related tests. The idea is that such tests may provide improved diagnostic sensitivity and quicker time to diagnosis. Such an approach has become more of a possibility as the costs for such testing is now little more than that previously set for sequencing a single gene. In this model, a clinician may send for such a panel after initial blood studies, if clinical suspicion is warranted, only going back to muscle biopsy and other tests if the genetic diagnosis was inconclusive.3
Dr. Tarnopolsky believes it is unwise to bring in such tests too early in the diagnostic process of suspected metabolic myopathies or muscular dystrophy, preferring to wait until other tests have helped rule in or out suspected disorders.
Dr. Finsterer suggests that beginning with the older, more conventional workup may sometimes be preferable for a patient with a phenotype that clearly suggests a particular disorder.
Dr. Tarnopolsky points out commercially available genetic tests often check only the exons of genes, and some patients may have deficits due to intronic mutations or a gene not yet assigned to a given disease. This can lead to false negative testing.
Dr. Finsterer agrees: “Even after application of next-generation technology for genetic work-up, a number of disorders may remain undetected.”

