
AJPhoto / Science Source
When evaluating patients with possible myopathic symptoms, rheumatologists must consider a rare, but important, group of inherited disorders: the metabolic myopathies. However, their diagnosis often remains a challenge. Early recognition of these primary metabolic myopathies is essential to help prevent disease morbidity and mortality from rhabdomyolysis. Here, we focus on the metabolic myopathies that present with primarily non-systemic issues and lead to exercise intolerance with dynamic myopathic symptoms.
Pathophysiology of Metabolic Myopathies
In cells, glucose and free fatty acids provide the main fuels used to produce adenosine triphosphate (ATP), the cell’s main source of energy. The process of glycogenolysis breaks down stored glycogen into a major source of usable
glucose. Through the processes of glycolysis, the Krebs cycle and, ultimately, the mitochondrial respiratory chain, the energy in glucose (4 kcal/g) and in free fatty acids (9 kcal/g) is liberated.1,2
However, some types of mutations lead to partial enzymatic function resulting in milder presentations. People with these types of mutations may have myopathic symptoms only, typically (though not always) non-fixed symptoms that occur in response to exercise or other metabolic stress. Such myopathies can prove challenging diagnostically, revealing exercise intolerance only without any other systemic abnormalities.1-4
Subtypes of Metabolic Myopathies
Metabolic myopathies are generally classified into three main subtypes: defects of glycogen metabolism, disorders of lipid metabolism, and mitochondrial disease.
Glycogen storage diseases

Dr. Tarnopolsky
Impairments in glycogenolysis, glycogen production or of anaerobic glycolysis are often grouped together as the glycogen storage diseases (GSDs). If not enough ATP can be made available to cells via glycolysis and glycogenolysis during intense exercise, cramping and other such symptoms can result.4
Mark Tarnopolsky, MD, PhD, FRCPC, is a professor of pediatrics and medicine in the Division of Neuromuscular and Neurometabolic Disorders at McMaster University, Hamilton, Ontario. He explains that we use specific energy sources during different types of activity: “If you had to go up a flight of stairs or run to catch the bus, the muscle is utilizing mostly glucose or glycogen. If there is glycogen storage disease, then this energy can’t be delivered to the muscle.”
Around half of the more than 20 currently known GSDs manifest with myopathic disease. The most common disorder of glycogen metabolism is also the most common metabolic myopathy overall: McArdle’s disease (GSD-V), whose prevalence is approximately one in 40,000.5
In a minority of disorders, symptoms arise not from problems with the lack of availability of products used for cellular metabolism, but because of structural changes due to the disease process itself. For example, in Pompe disease (GSD-II), the pathology derives from a lysosomal defect that leads to glycogen buildup in the lysosomes. This eventually damages the muscles, resulting in a fixed, progressive myopathy.
Fatty acid oxidation defects
Disorders of fatty acid oxidation and oxidation transport are less common than those of glycogen metabolism. Milder cases of such disorders can also present in adulthood as myalgia and exercise intolerance.6 Of the 18 or so identified genetic defects known to cause disease, around half are thought to cause myopathic symptoms. Carnitine palmitoyltransferase II (CPT II) deficiency is the most common disorder in this group.5
“Longer duration and lower intensity activities rely more heavily on lipid metabolism. These activities can precipitate an energy crisis in a fatty acid oxidation defect [FAOD] or mitochondrial disease,” explains Dr. Tarnopolsky. “[Patients] may [have] symptoms if they went for a two-hour walk and forgot to eat beforehand, if they rented bikes and went for a three-hour bike ride, that sort of stress. Eventually we run out of glycogen in our muscles and start dipping into fat stores.” This starts symptoms in these patients.
Mitochondrial myopathies
Disorders of the mitochondrial chain represent the third group of metabolic myopathies, resulting from problems with the final common pathway for energy derivation. These will often present with similar triggers as FAODs. However, most mitochondrial diseases will cause additional non-myopathic symptoms, as well. Depending on the mutation and severity, these can include diabetes, seizures, hearing loss, stroke and a wide variety of possible neurological symptoms. About one in 8,000 people are affected.1,5
Next generation sequencing via a rhabdomyolysis gene panel may be a more appropriate investigation than traditional muscle biopsy & related tests. The idea is that such tests may provide improved diagnostic sensitivity & quicker time to diagnosis.
Importance of Prompt Diagnosis
Although those with more severe infantile presentations will attract immediate investigation, people with milder phenotypes may experience symptoms for years before coming to medical attention. For example, many patients with McArdle’s disease are misdiagnosed with “growing pains” as children before they finally receive a correct diagnosis, often years later.6
Under ideal circumstances, metabolic myopathies should be diagnosed promptly. In a few cases, targeted therapies are available. In all cases, dietary and exercise interventions can help prevent morbidity and mortality from possible rhabdomyolysis, which can occur in these patients under conditions of metabolic stress. Rheumatologists can play an important role in diagnosing such patients and directing them to appropriate care.
Josef Finsterer, MD, PhD, is a professor of neurology at the University of Vienna, Austria. He points out that correct diagnosis can have important implications for the management of surgery and general anesthesia in such patients. Having the correct diagnosis also has implications for pregnancy, both for family planning purposes (and possible prevention of disease transmission) as well as for optimal management of pregnancy and delivery. Such conditions also need prompt diagnosis, since in some cases they are accompanied by serious medical problems that require management (especially in the case of mitochondrial disease). Accurate and early diagnosis can also prevent inappropriate and unnecessary interventions.1,5
Diagnosing Metabolic Myopathies
Dr. Tarnopolsky speculates that a rheumatologist may be most likely to encounter a patient with a metabolic myopathy as a referral from a family practice doctor for muscle weakness or for a high creatinine kinase (CK) level occurring with or without actual rhabdomyolysis.
“The challenge is that such a differential is extremely large,” he notes. This differential includes fixed structural genetic myopathies, such as limb girdle dystrophy, as well as inflammatory myopathies, such as dermatomyositis. “It’s sometimes challenging—separating those with metabolic disease from those with fixed structural disease or inflammatory disease and getting a final diagnosis—because there can be many mimics.”
Depending on the context, rheumatologists may need to consider other possible etiologies, such as endocrine or electrolyte-related myopathy, infectious myositis, malignant hyperthermia, other rare, genetic causes of myositis, medication-
related myositis, or non-myopathic syndromes, such as fibromyalgia.7
Medical history
As with many conditions in rheumatology, a detailed and careful medical history is critical for correct diagnosis of metabolic myopathies. Dr. Finsterer suggests dividing the history into two parts: undirected free speech of the patient followed by a structured questionnaire geared to recognizing possible key manifestations of myopathies.
Asking about exertional pain is key, because metabolic myopathic patients have usually suffered from exercise intolerance for years, with such symptoms as premature fatigue, episodic aches and cramps during activity. “When you ask them the questions, such as, ‘When you play soccer or hockey, were you having cramps? Did you ever get dark urine after intense exercise?’, often there is a positive history,” explains Dr. Tarnopolsky. “That’s different from a patient with inflammatory myopathy who usually has a subacute onset of progressive weakness, with or without a fixed myalgia.”
Unfortunately, getting such a history sometimes proves surprisingly difficult. Some patients may be so accustomed and adapted to their symptoms that they may deny exercise intolerance even if asked repeatedly. Yet after a correct diagnosis is made, they may retrospectively acknowledge their symptoms. It is sometimes helpful to ask family members about such complaints.
In people with fat oxidation defects, symptoms don’t usually occur unless people haven’t eaten in a long period of time or they engage in a longer duration activity. Other potential triggers can include extremes of temperature, general anesthesia and certain drugs (e.g., ibuprofen and diazepam).5
Dr. Tarnopolsky notes that some cases of FAOD or mitochondrial disease are picked up when a child gets sick and experiences sore muscles and/or rhabdomyolysis. He adds, “We do have a few, especially females, who may have mild symptoms. The patient might have worked out on the weekend, and then they go to the family doctor, have a high CK and might be sent to a rheumatologist.”
A diagnostic challenge: Even healthy people can sometimes overexert themselves, producing a high CK and even rhabdomyolysis. But the metabolic myopathy patient will usually have a recurrent history, experiencing symptoms from types of activities that would not give others such problems.
Some patients with mitochondrial disease present with a classic-appearing metabolic myopathy with episodic myopathic symptoms only. These overlap with many of the triggers of symptoms for FAOD disorders: prolonged exercise, fasting, fever, surgery or other such metabolic stressors. However, most patients with mitochondrial disease also present with non-myopathic symptoms that yield other diagnostic clues, and some will have a fixed, non-dynamic myopathy.5
Asking specifically about the second-wind phenomenon can be a critical clue for diagnosing McArdle’s disease (a glycogenolytic defect). Because of the particular nature of the deficit, McArdle’s patients often report a “second wind phenomenon” due to the delivery of blood-borne substrates to the muscle. A typical history is that the initial cramping sensation with vigorous exercise may lessen after a short rest or reduction in intensity, followed by a resumption of activity that feels much easier. Patients with GSDs with glycolytic-type defects do not experience a “second wind.”4
History is also key in distinguishing a possible metabolic myopathy from an inflammatory myopathy or a muscular dystrophy. “Generally, someone with an inflammatory myopathy is going to have a subacute presentation,” Dr. Tarnopolsky notes. “Thus, it’s going to be weeks to months when they experience the weakness, and it’s usually insidious in its onset.” He notes that muscular dystrophies can look very similar, though they usually have a much longer time of gradual onset.
A comprehensive review of systems is also warranted, with emphasis on potential symptoms that might point to particular metabolic myopathies or other myopathy types (see Table 1).
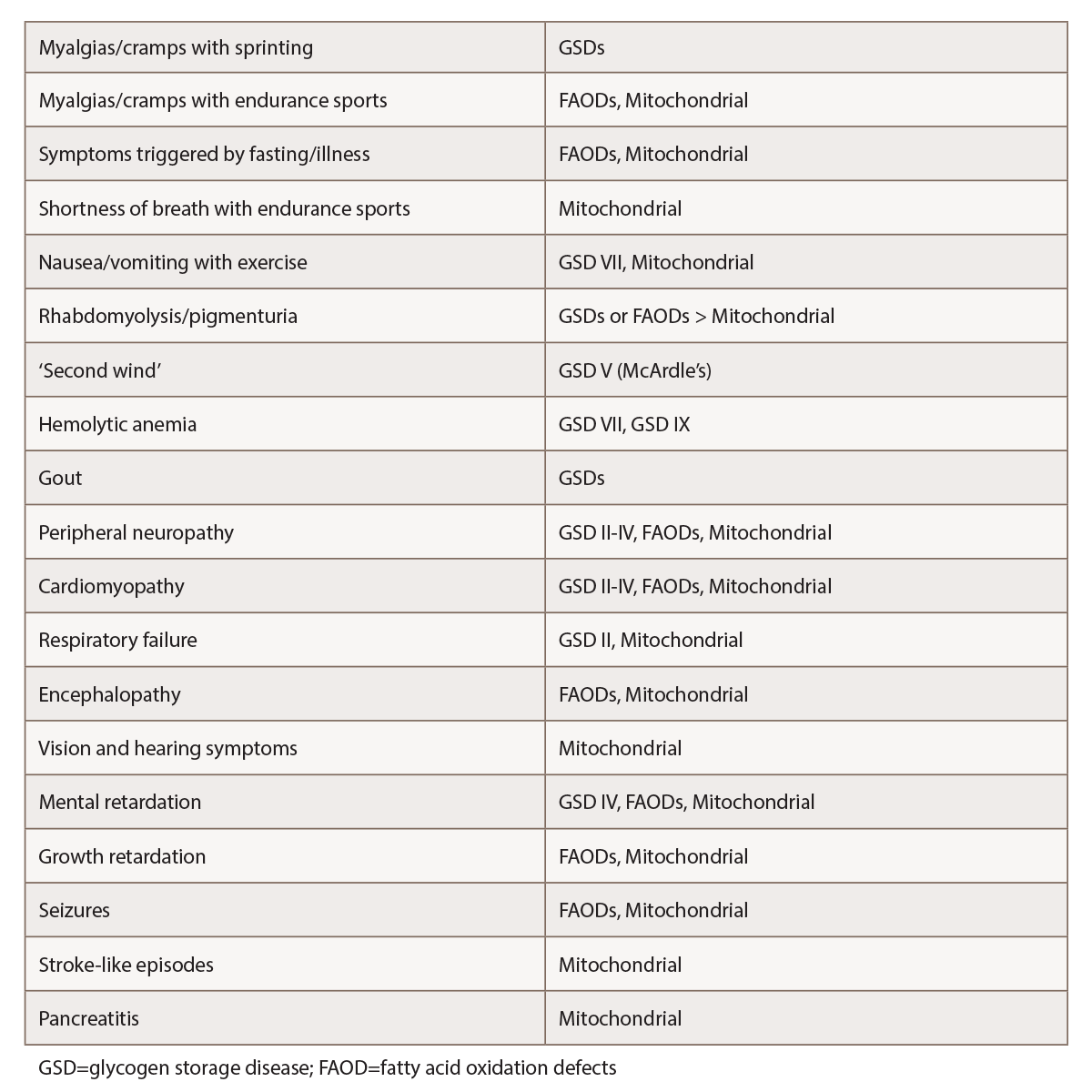
(click for larger image) Table 1: Selected Features Suggestive of Metabolic Myopathies1,2,4,5
(may have myopathic symptoms only)
The physical
The physical also plays a foundational diagnostic role for these patients. “This includes a proper neurological examination with mental status, cranial nerves, motor, reflexes, sensory and gait,” says Dr. Tarnopolsky. A fixed, motor weakness may be present in cases of McArdle’s disease or mitochondrial disorders. In contrast, muscular dystrophy will typically present with obvious muscle weakness, although sometimes the only finding is exercise-induced cramps without other obvious clinical clues.6
Dr. Tarnopolsky explains that for people with fat oxidation defects and glycogen storage diseases, the physical exam is often completely normal (though if a patient has just experienced rhabdomyolysis, they may initially be achy and weak). In contrast, patients with muscular dystrophy or inflammatory myopathy are more likely to display symptoms like shoulder girdle weakness, winging of the scapula, difficulty abducting the arms and difficulty with hip flexion.
The joint and skin exam are also key in establishing or ruling out a potential inflammatory myopathy. “In the metabolic myopathies and the structural myopathies, you are not going to have your Gottron’s rash, your heliotrope rash, your shawl sign, your holster sign, the splinter hemorrhages—all the other things that usually accompany the inflammatory myopathies,” says Dr. Tarnopolsky.
Assessing for muscle atrophy and muscle hypertrophy is also helpful. “Becker and some of the limb girdle muscular dystrophies have enlarged calf muscles, and they are
usually quite strikingly enlarged; whereas, dysferlin and ANO5 type Miyoshi/limb girdle myopathies usually have very thin calf muscles,” says Dr. Tarnopolsky. He notes that muscle bulk changes, such as these, are much more likely to indicate a genetic structural myopathy than an inflammatory or a metabolic myopathy.
Some patients with mitochondrial disease will also have a fixed myopathy. “To help identify those cases, usually there are cranial nerve findings, such as optic atrophy, sensorineural hearing loss, ptosis and ophthalmoparesis, none of which are features of inflammatory myopathy.”
Initial laboratory clues
Guided by clues from the history and physical, initial basic lab work may include tests for CK, myoglobin, basic renal and liver function, lactate, urate, vitamin D, thyroid function, complete metabolic panel (CMP), aldolase, anti-nuclear antibodies, fasting acylcarnitine profile and urine organic acids, as well as tests for inflammation and infection.3,5,7 Dr. Finsterer notes that reviewing all previous tests can be challenging, but is an essential part of the diagnostic process.
Dr. Tarnopolsky recommends the CK as an inexpensive and simple test that can begin to sort out a fixed genetic structural myopathy from an inflammatory, metabolic or other kind of myopathy. “Usually, with glycogen storage disease and [FAOD] patients, the CK is going to go up with metabolic stress, and it usually comes back to normal,” explains Dr. Tarnopolsky. In contrast, CK findings from someone with a structural myopathy such as Becker or limb girdle will show a consistently elevated CK. He says, “If you follow the CK, it stays pretty similarly elevated; so if it’s 2,000 one time, it’s 2,300 the next time.” An inflammatory myopathy, if left untreated, will often show a CK that continues to go up over time.
One exception to this general pattern is the McArdle’s disease patient. “Unlike many metabolic myopathies, where the CK goes up with stress and comes back down to normal, the McArdle’s patient’s CK often stays persistently elevated. And that’s why it can look more like an inflammatory disease or muscular dystrophy,” Dr. Tarnopolsky explains. These patients may also be unusual in that they may not present with the classic exercise-induced symptoms and may appear to be chronically weak, with only minimal activity-induced symptoms.
Dr. Tarnopolsky says a blood acylcarnitine test can be particularly helpful if someone has symptoms induced by fasting or exercise and a FAOD is suspected. As he notes, “A normal acylcarnitine provides pretty good evidence that you have ruled out a fatty oxidation defect.”
He also notes that a fasting lactate can be helpful if a rheumatologist notes anything suspicious on the neurological exam, because an elevated lactate may indicate a mitochondrial myopathy. “If for example, you saw optic atrophy, ptosis, ophthalmoparesis on your exam and you are thinking mitochondrial disease, we would also add plasma amino acid testing. About 20% of [mitochondrial] patients will have an elevated alanine.” He says a metabolic screen on the urine, including metabolic acids, is also indicated in such a situation, because there is often an increase in 3-methylglutaconic acid.
If an inflammatory myositis is still in the differential, Dr. Tarnopolsky suggests a myositis panel. “These are reasonably sensitive and specific. If you had a positive Jo-1 antibody or one of the other classic markers for inflammatory myopathy, it would be very unlikely for that to occur in a metabolic myopathy or structural myopathy.”
Second-line investigations
Follow-up second-line studies depend on previous findings, clinical preference and institutional availability of certain tests. Historically, the next move would typically be investigation via electromyography (EMG) or nerve conduction studies, both of which we would expect to be essentially normal for glycogen storage disease, fat oxidation disorders and most mitochondrial disorders. EMG is often abnormal in muscular dystrophy and inflammatory myopathies. A muscle biopsy is often the next potential test, providing important diagnostic clues regarding potential metabolic myopathies, genetic structural myopathies or inflammatory myopathies.
However, some have suggested moving genetic testing to an earlier point in the process, at least in some instances. For example, some have argued that next-generation sequencing via a rhabdomyolysis gene panel may be more appropriate than traditional muscle biopsy and related tests. The idea is that such tests may provide improved diagnostic sensitivity and quicker time to diagnosis. Such an approach has become more of a possibility as the costs for such testing is now little more than that previously set for sequencing a single gene. In this model, a clinician may send for such a panel after initial blood studies, if clinical suspicion is warranted, only going back to muscle biopsy and other tests if the genetic diagnosis was inconclusive.3
Dr. Tarnopolsky believes it is unwise to bring in such tests too early in the diagnostic process of suspected metabolic myopathies or muscular dystrophy, preferring to wait until other tests have helped rule in or out suspected disorders.
Dr. Finsterer suggests that beginning with the older, more conventional workup may sometimes be preferable for a patient with a phenotype that clearly suggests a particular disorder.
Dr. Tarnopolsky points out commercially available genetic tests often check only the exons of genes, and some patients may have deficits due to intronic mutations or a gene not yet assigned to a given disease. This can lead to false negative testing.
Dr. Finsterer agrees: “Even after application of next-generation technology for genetic work-up, a number of disorders may remain undetected.”
Referral & Ongoing Care
Although rheumatologists are typically comfortable with the diagnosis and treatment of inflammatory myopathies, if a metabolic myopathy is suspected, referral to a metabolic neurologist is indicated. There, more specialized diagnostic tests can be performed. If a rheumatologist has strong clinical suspicion of a structural myopathy, referral to a neuromuscular neurologist would also be prudent. In both cases, these specialists may be better suited performing some of the specific, second-line tests involved in diagnosing these rare conditions.
Dr. Tarnopolsky recommends patients diagnosed with metabolic myopathies be treated at a specialty center in a major academic institution, because the treatment is highly disease specific, and new treatments are always emerging. A few of the metabolic myopathies have established specific treatments, such as enzyme replacement therapy for Pompe disease or riboflavin for MADD (multiple acyl-CoA dehydrogenase deficiency). Specially tailored exercise therapy also seems to benefit many patients, and depending on the condition, specific dietary plans can help.4,5
But for the most part, treatment centers on helping patients avoid disease triggers and prevent rhabdomyolysis. Rheumatologists can play an important role in identifying the patients at risk of this potential serious complication due to an undiagnosed metabolic myopathy.
Ruth Jessen Hickman, MD, is a graduate of the Indiana University School of Medicine. She is a freelance medical and science writer living in Bloomington, Ind.
References
- van Adel BA, Tarnopolsky MA. Metabolic myopathies: Update 2009. J Clin Neuromuscul Dis. 2009 Mar;10(3):97–121.
- Berardo A, DiMauro S, Hirano M. A diagnostic algorithm for metabolic myopathies. Curr Neurol Neurosci Rep. 2010 Mar;10(2):118–126.
- Olpin SE, Murphy E, Kirk RJ, et al. The investigation and management of metabolic myopathies. J Clin Pathol. 2015 Jun;68(6):410–417.
- Tarnopolsky MA. Metabolic myopathies. Continuum (Minneap Minn). 2016 Dec;22(6, Muscle and Neuromuscular Junction Disorders):1829–1851.
- Finsterer J. An update on diagnosis and therapy of metabolic myopathies. Expert Rev Neurother. 2018 Dec;18(12):933–943.
- Quinlivan R, Jungbluth H. Myopathic causes of exercise intolerance with rhabdomyolysis. Dev Med Child Neurol. 2012 Oct;54(10):886–891.
- Smith BW, McCarthy JC, Dawley CA. Suspect myopathy? Take this approach to the work-up. J Fam Pract. 2014 Nov;63(11):631–638.

