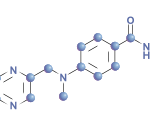
In ancient Roman mythology, the two-faced Roman God Janus presided over all beginnings and transitions. On November 6, 2012, the U.S. Food and Drug Administration (FDA) approval of the first Janus kinase (JAK) inhibitor, tofacitinib, for treatment of rheumatoid arthritis (RA) heralded the advent of kinase inhibitors in the therapeutics of rheumatic diseases. Despite the advances in treatment of RA over the last two decades by the use of cytokine and target cell inhibitors, a significant percentage of RA patients either do not respond or lose their response to therapy. The pathophysiology of rheumatoid arthritis is dependent on the activation of both the innate and adaptive immune systems. The cells that comprise the immune system utilize kinases as the gatekeepers to determine how they should respond to external stimuli such as antigens, cytokines, and immune complexes. The use of new small molecule inhibitors against cytoplasmic kinases such as the JAK family, splenic tyrosine kinase (SYK), or mitogen-activated phosphokinase p38 (p38MAPK) will alter the resulting dysregulated immune systems in different ways and hopefully further promote our understanding of its complexities.