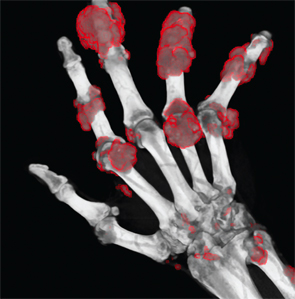
Figure 2: DECT of hands #1
Pegloticase is a new alternative therapy for patients with severe, refractory gout unresponsive to other urate-lowering agents. The goal of this therapy is to reduce disease burden, tophi size and frequency of flares and to improve quality of life when other treatments have failed. Persistent lowering of plasma uric acid (PUA) to less than 6 mg/dL, while achieving the therapy’s goal, is considered successful treatment.
Prior studies have shown that although all patients initially respond to pegloticase, as indicated by decreased PUA levels, not all patients maintain that response. The reasons are multifactorial, but as research has progressed, several studies have shown that gout can be heritable, and both genetics and antibody formation to drug therapy can affect both overall disease and response to treatment.
Herein, we present a case of severe refractory gout in a patient unresponsive to all standard urate-lowering therapies, as well as treatment with pegloticase.
Case Description
A 65-year-old African American male with a past medical history of stage II chronic kidney disease and hypertension was referred to our clinic for poorly controlled, erosive, polyarticular gout. The patient was first diagnosed at the age of 45 with initial joint involvement noted only in the right knee and left wrist. Over time, he had gradual progression of his disease, with involvement of his hands, wrists, elbows, knees, ankles and feet, as well as development of multiple tophi over his extremities. Moreover, he had experienced numerous flares over the course of his disease, averaging approximately six each year.
(click for larger image)
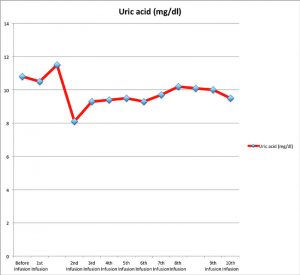
Figure 1: Uric acid (mg/dL) variation prior to and at the time of each pegloticase infusion
At the age of 55, 10 years after the onset of his gout, he required bilateral total knee replacements due to the severity of his disease. Over the past five years, he has had multiple tophi removed from his distal interphalangeal joints (DIPs), elbows and ankles in an effort to improve his overall function. Unfortunately, tophi reaccumulated after each procedure.
At the time of his initial visit to our clinic, he had already failed multiple treatments with non-steroidal anti-inflammatory drugs, colchicine, probenecid, allopurinol and several rounds of prednisone. Given his severe condition, the patient had a significantly decreased quality of life, because he was disabled by the extent of his disease.
Physical examination revealed multiple tophi over his right ear pinna, right third metacarpophalangeal (MCP), right fifth proximal interphalangeal (PIP), left third and fourth DIP, bilateral elbows, bilateral ankles and left foot involving the second and third metatarsophalangeal (MTP) joints. Range of motion was severely limited due to the presence of his numerous tophi.
Laboratory analysis revealed an elevated creatinine to 1.7 and PUA of 9.6 mg/dL.
Hand X-rays from three years prior were notable for severe polyarticular erosive disease. Plain films obtained in our institution confirmed multiple erosions involving the MCP, DIP and PIP joints, overlying soft tissue masses containing amorphous calcifications, loss of cartilage in the intercarpal bones, and carpal bony erosions. Plain films of the elbows and ankles similarly revealed large amorphous deposits and calcifications. Multiple erosions were also present at the first metatarsal head, tarsometatarsal joints and at the bases of the metatarsal and intertarsal joints.
(click for larger image)
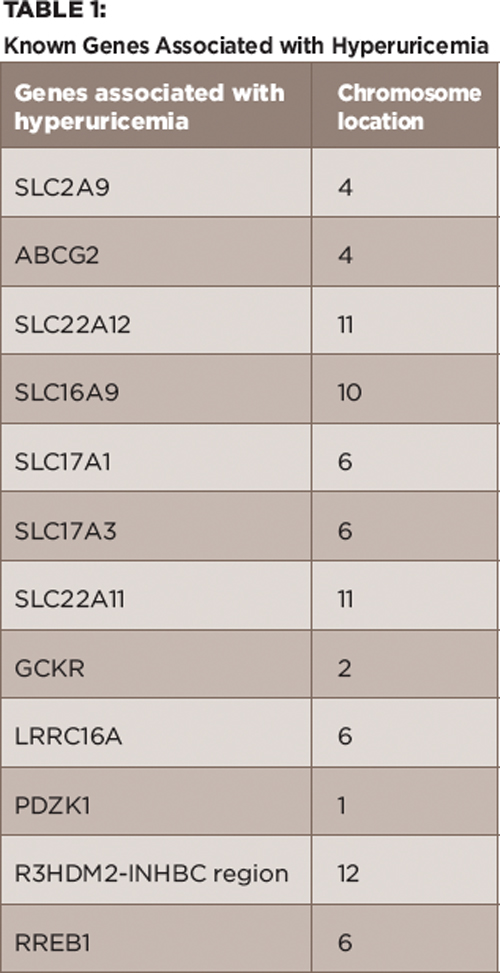
Table 1:
Known Genes Associated with Hyperuricemia
At that time, treatment with febuxostat was initiated at 40 mg/day, which was gradually increased to 120 mg/day with limited results. Initially, the patient noted a slight improvement in the frequency of his gout flares, but soon he began having recurrent flares, as before. He started to require frequent prednisone bursts and, ultimately, ended up using low-dose prednisone as prophylaxis. His PUA was monitored every three months during this period, and he remained in the range of 9–10 mg/dL.
Given his treatment failure with febuxostat, the decision was made to start pegloticase. Prior to initiating this treatment, a new set of plain films was obtained, which showed progression of tophi, as well as new erosions in multiple sites involving his hands, elbows, feet and ankles.
Pegloticase was administered intravenously at 8 mg every two weeks for a total of 10 infusions. In addition, he was treated with 20 mg of prednisone daily because he continued to have acute gout flare-ups on lower doses of prednisone. PUA was checked before each infusion, and the values are reported in Figure 1. After a few infusions, based on the reported PUA values, we discussed with the patient the possibility of discontinuing the infusion due to fear of an infusion reaction from antibody formation. The patient, however, accepted the risks and decided to continue the infusion.
Over the course of his five months of infusions, he subjectively reported improvement in his range of motion, as well as a decrease in the size of his tophi. However, clinically these findings were not appreciated. And although the frequency of gout flares was reduced, this was likely secondary to the prednisone more than the pegloticase itself. Unfortunately, with the exception of one value of 8.1 mg/dL at the time of the second infusion, PUA values remained elevated in his pretreatment range of 9–10 mg/dL. Overall, the infusions were well tolerated, and no hypersensitivity reactions occurred; however, we were unable to check if the patient developed antibodies against pegloticase.
Following treatment with pegloticase, dual-energy CT images were obtained, and the images were compared with those done the prior year. Beyond the multiple erosions, the images showed extensive tophi and deposition in the flexor and extensor tendons of hands, ankles and feet (see Figure 2 and Figures 3–5). Based on the persistently elevated PUA levels (with no value ever recorded less than 6 mg/dL) and the unchanged tophi, our patient was deemed to have failed pegloticase, and the treatment was stopped after approximately five months.
Discussion
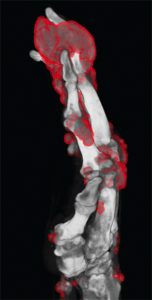
Figure 3: DECT of hands #2
Pegloticase is a mammalian PEGylated recombinant form of uricase that catalyzes the oxidation of uric acid to allantoin and is used in cases of severe refractory gout. In prior studies, it has been shown to significantly lower PUA and reduce tophi burden by more than 80% in those who were able to maintain PUA less than 6 mg/dL.1-2 However, not all patients treated with pegloticase maintain their response, and some begin to lose their response, leading to increased PUA and infusion reactions. This can be due to a variety of reasons, including antibody formation, as well as genetic predisposition and abnormalities in the renal urate transport system.
It should be noted that in initial and subsequent studies, all patients given pegloticase initially had significant decreases in their PUA.2-4 In a study by Sundy et al, it was noted that nonresponders in the bimonthly treatment groups still had a mean PUA of less than 6 mg/dL until Week 10 and in the monthly treatment group until Week 5.2 In our particular case, the patient never once obtained a PUA of less than 6 at any point during his five-month treatment course (lowest recorded value was 8.1 mg/dL) or had any clinically detectable decrease in tophi burden.
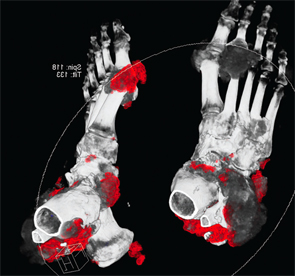
Figure 4: DECT of feet #1
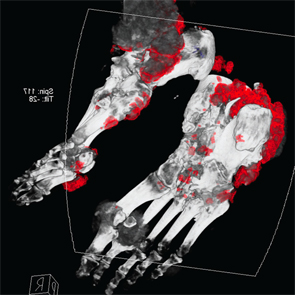
Figure 5: DECT of feet #2
One known reason for failure of pegloticase and decreased response over time is antibody production. Three known antibodies have been described in the literature: anti-PEG, anti-pegloticase and anti-uricase.
In a study by Lipsky et al, it was shown that these antibodies play a clinically significant role in pegloticase clearance and that 89% of patients had anti-pegloticase antibodies after the first infusion.4 However, in those who responded to pegloticase (defined by PUA level less than 6 mg/dL for 80% or more of the three- to six-month study period), they typically had low titers of the antibody (<1:2,430). Additionally, during the first two months of treatment, responders showed a tendency toward formation of IgM antibodies and not IgG, whereas nonresponders typically had IgG formation by Week 3, as well as high titers of anti-pegloticase antibodies.
Lastly, the study noted that in those who lost their response, a correlation was seen between higher antibody titers and rise in PUA, as well as increased infusion reactions as PUA began to rise. Nonetheless, it should be mentioned that again in this study all patients responded initially to pegloticase with a PUA of less than 6 mg/dL with the mean loss of response time being six weeks. Further, although this may have had some role in our patient’s lack of response, it is not fully explained by this, nor did he ever develop any sort of infusion reaction, as seen in those with high titers of antibodies.
Genetics have been shown to have an increasing role in hyperuricemia and gout. Based on several genome-wide association studies, numerous genes are known to be involved in the pathogenesis of hyperuricemia, most of which involve the uric acid transportasome.
The uric acid transportasome consists of a group of proteins that are coded by these particular genes as part of the renal urate transport system. There are several components of the transportasome each with multiple variants and single nucleotide polymorphisms that can lead to hyperuricemia.5 Known variants are included in Table 1. Each gene and their variants can lead to hyperuricemia in different ways through renal overload or renal under-excretion. For example, SLC2A9 codes for a glucose transporter (GLUT-9), which is involved in the reabsorption of filtered urate in the proximal tubules. In most genomic studies, this has been the most statistically significant genetic determinant of PUA.5 A loss of function mutation in this gene leads to renal hypouricemia.
Other studies have also shown that abnormal expression of GLUT-9 in articular chondrocytes may play a role in uric acid crystal deposition as well.6 Several more examples exist; however, these genomic studies have also shown variability between different ethnicities. In particular, for African Americans, mutations in ABCG2 and SLC22A12, two known genetic loci for hyperuricemia, may play an even larger role.7,8
A study done by Tin et al looking specifically at genetic-wide associations for African Americans showed a novel variant of SLC22A12, which codes for URAT1, that has an essential role in urate transport in the proximal tubules through the apical absorption of urate. This variant was shown to lead to substantially reduced urate transport.
Finally, variants of ABCG2 (an ATP binding cassette protein) have been shown to have a large effect on PUA in both men and African Americans.5,7 In particular, a single nucleotide polymorphism, which leads to a Glu131Lys amino acid substitution, has been shown to cause a 53% reduction in the rate of ABCG2-mediated urate transport.5
In conclusion, both genetics and antibody formation can lead to treatment failure. However, to our knowledge this is the first reported case of complete pegloticase failure in which reduction in PUA was never obtained at any point during treatment. The likely reason for this may be an underlying genetic predisposition, potentially in one of the renal-urate transporters, which led to multiple treatment failures in this patient.
As genomic research and our understanding of the pathophysiology of gout progress, further targets for drug development are being identified.9 One such advance is the development of lesinurad, which targets URAT1, and will hopefully lead to more potential treatment options for such patients as these in the future.

Diana M. Girnita, MD, PhD, is a rheumatology fellow at the University of Cincinnati in the Department of Immunology, Allergy and Rheumatology.
Cody Lee, MD, is a third-year internal medicine resident at the University of Cincinnati in the Department of Internal Medicine.
Christine Chhakchhuak, MD, is an assistant professor at the University of Cincinnati, in the Department of Immunology, Allergy and Rheumatology. She is board certified in rheumatology and internal medicine.
References
- Baraf HS, Becker MA, Gutierrez-urena SR, et al. Tophus burden reduction with pegloticase: Results from Phase 3 randomized trials and open-label extension in patients with chronic gout refractory to conventional therapy. Arthritis Res Ther. 2013 Sep 26;15(5):R137.
- Sundy JS, Baraf HS, Yood RA, et al. Efficacy and tolerability of pegloticase for the treatment of chronic gout in patients refractory to conventional treatment: Two randomized controlled trials. JAMA. 2011 Aug 17;306(7):711–720.
- Hershfield MS, Ganson NJ, Kelly SJ, et al. Induced and pre-existing anti-polyethylene glycol antibody in a trial of every 3-week dosing of pegloticase for refractory gout, including in organ transplant recipients. Arthritis Res Ther. 2014 Mar 7;16(2):R63.
- Lipsky PE, Calabrese LH, Kavanaugh A, et al. Pegloticase immunogenicity: The relationship between efficacy and antibody development in patients treated for refractory chronic gout. Arthritis Res Ther. 2014 Mar 4;16(2):R60.
- Reginato AM, Mount DB, Yang I, et al. The genetics of hyperuricaemia and gout. Nat Rev Rheumatol. 2012 Oct;8(10):610–621.
- Vitart V, Rudan I, Hayward C, et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet. 2008 Apr;40(4):437–442.
- Zhang L, Spencer KL, Voruganti VS, et al. Association of functional polymorphism rs2231142 (Q141K) in the ABCG2 gene with serum uric acid and gout in 4 US populations: The PAGE Study. Am J Epidemiol. 2013 May 1;177(9):923–932.
- Tin A, Woodward OM, Kao WH, et al. Genome-wide association study for serum urate concentrations and gout among African Americans identifies genomic risk loci and a novel URAT1 loss-of-function allele. Hum Mol Genet. 2011 Oct 15;20(20):4056–4068.
- Diaz-torné C, Perez-herrero N, Perez-ruiz F. New medications in development for the treatment of hyperuricemia of gout. Curr Opin Rheumatol. 2015 Mar;27(2):164–169.


