Though I have labored in the rheumatologic vineyards as a clinician for more than 35 years, the details of my first cases of polymyalgia rheumatica (PMR) and giant cell arteritis (GCA) are still fresh. The former brought a bottle of holiday cheer, with gratitude for the triumphant effects of low-dose prednisone on aching and stiffness of nearly disabling extent; the latter, nothing but heartache. On three occasions during the preceding month, the patient with GCA had hunted medical care for symptoms of anorexia, low-grade fevers, and occipital headaches, twice from her general practitioner and once from a local emergency room. When I first met her, she had just been hospitalized, having lost vision in the left eye the previous day. Now she was admitted to hospital with amaurosis fugax affecting the right eye, which, despite the use of high-dose corticosteroids, ended in permanent loss of sight in that eye as well. Since that first year of fellowship, both diseases remain at the core of my clinical interests, and a potential referral for either still quickens the clinical pulse.
PMR and GCA: Relatedness, Including Misnomers
The kinship of PMR and GCA is well known. PMR is found in up to one-half of patients with GCA (though the two need not occur synchronously). GCA is commonly cited as affecting 15% of patients with PMR, though in clinical practice that percentage is probably somewhat lower. Both are quintessential conditions of older adults—in effect, never occurring under the age of 50, with 90% of cases over the age of 60, and a median age of onset in the 70s. The clinical and epidemiologic connections carry down to the immunologic level, as the temporal arteries from patients with PMR and GCA both contain activated dendritic cells and similar cytokine profiles—with the notable exception of interferon-γ (IFN-γ), which is present in the temporal arteries of patients with GCA but absent in PMR, and which likely plays a pivotal role in the transformation from a vasculitis that is subclinical to one that is clinically explicit.1
PMR: Treatment Needs a Clinician, Not a Logician
However misleading the name, the clinical presentation of PMR is often immediately appreciated. Many clinicians will recognize a message from their front office to the effect that, “Mrs. A’s daughter called to ask if there is any way her mother’s appointment can be moved up—says that for the past two weeks she is so stiff she can hardly get out of bed in the morning.” Though the onset of the symptoms of PMR may be subacute, more often than not it is abrupt, occasionally startlingly so—patients have told me they went to bed feeling good, but awakened in the morning as though run over by the proverbial truck. The rapid onset of symptoms suggests the possibility of an exposure, particularly to an infectious agent, but the search for evidence of such a pathogen in PMR has been, to date, unrevealing. I have also been struck by what seems to be a curiously frequent reference to unusual physical activity just prior to the onset of symptoms of PMR—though I acknowledge the common penchant of patients to ascribe any musculoskeletal symptoms to prior trauma or exertion.
The ferocious quality of the gel phenomenon in PMR is also distinctive. Though all systemic rheumatic diseases with synovitis are marked by gelling with inactivity, this symptom is particularly prominent in patients with PMR. Waking up at night with the mere act of rolling over in bed, needing help from a spouse with morning activities of daily living, especially with upper- and lower-extremity dressing, trouble transferring out of a car seat after a forty-five-minute ride—all testify to the potency of such gelling.
That such a symptom is swiftly—and often spectacularly—abolished with low-dose corticosteroids is a distinguishing hallmark of PMR (See Figures 1 and 2, at right). That such a response could not be validated as a criterion that discriminates PMR from comparator diseases in the recently published “2012 Provisional Classification Criteria for Polymylagia Rheumatica” is surprising.5 Granted that these criteria are for classification, not diagnosis, the fact remains that the brisk therapeutic response to low-dose corticosteroids is clinically remarkable. It is has been argued that the use of this response for diagnosis of PMR is an exercise in tautology; I’m no logician, but I believe that, until we have a reliable biomarker (or biomarkers) for PMR, clinicians will continue to assess corticosteroid responsiveness in the diagnosis of a patient with suspected PMR.6


PMR: Troubles with the Taper
The initial treatment of the patient with PMR is the easy part; the hard part can be the subsequent treatment. Even at low doses, the side effects of corticosteroids (CS) can be formidable, such as the well-recognized bone loss, and also ones less documented in the usual litany of toxicities, such as widespread capillary fragility—not an uncommon problem in the older adult on concurrent treatment with aspirin, warfarin, or other anticoagulants. Furthermore, the need for prolonged treatment with CS in the treatment of PMR is not uncommon; relapses during the CS taper are usual, rather than exceptional. Why PMR is monophasic in some patients and polyphasic in others—with recurrences seen even years after CS have been discontinued—is unknown.7 The benefit of steroid-sparing medications in the taper of CS—viz., methotrexate—is unfortunately marginal, so the goal of treatment centers on identifying the lowest dose of CS that keeps the given individual patient comfortable, with periodic efforts to accomplish a complete taper of the drug. Owing to the exquisite sensitivity to CS exhibited by many patients with PMR, the use of 1-mg decrements may facilitate this process.
The taper of CS can be problematic for several reasons. It may not be a problem per se, but simply part and parcel of the natural history of PMR, which, as noted above, is marked by relapses and recurrences. The diagnosis may have been just plain wrong—e.g., multifocal osteoarthritis or fibromyalgia may have been mistaken for PMR. Or, symptoms referable to commonplace local problems—nodal osteoarthritis, rotator cuff pathology, OA at the hips—may recrudesce during the taper of CS, which must be distinguished from bona fide flares of PMR. Or, signs of explicit synovitis may be recurrent, and raise consideration for so-called “late onset rheumatoid arthritis” or “elderly onset rheumatoid arthritis.”
Distal symptoms and signs can be identified in up to one-half of patients with PMR—carpal tunnel syndromes, distal arthralgias, and a modest peripheral arthritis, most common at metacarpophalangeal and wrist joints, as briskly responsive to low-dose CS as are the proximal symptoms, and never erosive. A positive anti-CCP antibody will flag a diagnosis of rheumatoid disease in a patient with a polymyalgic presentation and distal findings. But polyarthritis of abrupt onset in older adults is not uncommonly seronegative, and clinical and serologic attempts to differentiate PMR at presentation from something that is better construed as rheumatoid disease have not been particularly successful. Practically speaking, it may take three to six months of attentive follow-up to sort things out; if peripheral arthritis recurs with efforts to get the dose of prednisone below 10 mg per day in a patient initially thought to have PMR, the diagnosis—and therefore treatment—must be reassessed. A reliable biomarker for PMR—or set of such markers—would obviously be a boon.
Considering GCA
What PMR, a musculoskeletal problem with a special predilection for the synovium of the proximal joints and bursae, has to do with GCA, a systemic granulomatous arteritis, is a mystery. Their epidemiologic relationship has already been touched on. A striking feature of GCA is the heightened prevalence in Scandinavians, and its decidedly uncommon occurrence in African-Americans—a genetic clue that remains to be deciphered.8 Data for such prevalence in PMR are not available, but clinical experience suggests that both PMR and GCA are much more common in Northern Europeans than African-Americans and Latinos. That said, all ethnic and racial groups can be affected—indeed, my patient whose GCA went undiagnosed was of African origin, by way of Barbados and, before that, the slave trade from West Africa.
The Diagnosis of GCA
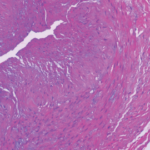
The diagnostic stakes in GCA are of course much higher than in PMR, because of the potential consequences of a missed diagnosis—loss of vision—and the potential toxicities of the treatment that ensues—customarily, high-dose CS. I remain of the opinion that GCA is not a clinical diagnosis, and that every effort should be made to premise this diagnosis on histopathological—or additional—footing. The traditional means of securing histopathological evidence is the humble temporal artery biopsy, which is minimally invasive, simple, and benign. However, the performance of the temporal artery biopsy as a diagnostic test is beset with, as my granddaughter puts it, issues. What length? One side? Both sides? Simultaneous? Sequential? My eminence-based take is that, in the appropriate clinical context, a unilateral, shorter segment biopsy has a high negative predictive value for the diagnosis of GCA—but it is unlikely that the merits and deficiencies of the temporal artery biopsy are amenable to definitive resolution.
Ultrasound examination of the temporal artery was proposed as a noninvasive assessment for GCA more than 15 years ago. Though earlier analyses expressed concerns about sensitivity and specificity, the technique has persisted, much more so in Europe than in the U.S. (another rheumatologic mystery), and recent studies continue to cite an adequate performance as a diagnostic test for GCA.9,10 Still, given the inescapable fact of its operator dependency, the diagnostic value of this procedure will continue to be controversial. A noninvasive, sensitive, and specific test for the diagnosis of GCA remains a major unmet need.
What about a strongly suspected diagnosis of GCA that cannot be confirmed by histopathology or other means? Most of us certainly treat such patients—though I find “biopsy-negative GCA” as among the most fraught of rheumatologic diagnoses. It might be noted that there is scant literature on these patients, and that most of the published studies on GCA—diagnosis, treatment, and clinical course—are largely composed of patients who are biopsy positive.
GCA: What’s Easy, What’s Not
What’s easy in GCA is its treatment, specifically, the prevention of blindness. Corticosteroids work. If there is no vision loss at the time that GCA is diagnosed, and if treatment is instituted with higher doses of CS, given daily, the risk of subsequent vision loss is very small, probably less than 1%.11 (If vision loss due to GCA has already occurred, a small percentage of patients may experience further deterioration of vision during the first week or two of treatment.) I am, of course, painting with a broad brush; there are manifold difficulties in the treatment of GCA, nearly all of which have to do with the toxicities of higher doses of CS. But the proven ability of the appropriate and timely administration of CS to prevent blindness in GCA is a yardstick against which any other treatment must be measured.
Could CS toxicities be lessened by using a lesser dose than the traditional 1 mg/kg? The dosing of CS for the treatment of GCA is based on retrospect and custom; there are no prospective studies. No doubt the specter of blindness drives the choice of high-dose CS. If patients could be stratified for the risk of blindness, perhaps the CS dose could be accordingly calibrated. No clinically reliable means for such stratification have emerged; probably the strongest risk factor for subsequent permanent loss of vision is a history of prior amaurosis. It is also worth bearing in mind that the majority of patients with GCA in fact do not incur the catastrophe of vision loss. Such was the case of the first reported GCA patient, one Mr. Rumbold, as vividly described by Hutchinson in 1890.12
“He had red streaks on his head which were painful and prevented him wearing his hat….The red streaks proved, on examination, to be his temporal arteries, which were on both sides found to be inflamed and swollen. The streaks extended from the temporal region almost to the middle of the scalp and several branches of each artery could be distinctly traced. The conditions were nearly symmetrical. During the first week that he was under my observation, pulsation was freely detected in the affected vessels, but it finally ceased; the redness then subsided, and the vessels were left impervious cords.”
Mr. Rumbold lived several years more, without vision loss or, apparently, other sequelae of GCA.
The contemporary management of GCA involves pharmacologic difficulties beyond the potential toxicities of CS. Should low-dose aspirin be used? Two retrospective studies found a protective effect against so-called cranial ischemic events (vision loss and stroke), two others did not.13-16 Is prophylaxis against pneumocystis pneumonia (PCP) warranted? Is the risk of PCP in fact increased in patients with GCA on monotherapy with high-dose CS? What is the risk–benefit ratio of bisphosphonates for the reduction of fracture risk? It is conceivable that a patient with newly diagnosed GCA could leave the office with prescriptions for high-dose prednisone, low-dose aspirin, a proton pump inhibitor (because of the increased risk of gastrointestinal bleeding in an older adult on concurrent CS and aspirin), trimethoprim-sulfamethoxazole (provided there is no sulfa allergy), and a bisphosphonate. For whatever it’s worth, in such a patient, I currently do not routinely deploy low-dose aspririn or pneumocystis pneumonia prophylaxis, as I am unpersuaded as to their value by either the published studies or clinical experience, but I do aggressively use bisphosphonates, which, despite their recently tarnished reputation, have been of clear value in decreasing fracture risk from CS.
And the management of GCA beyond its initial treatment carries a further set of difficulties. How quickly should CS be tapered, and how long should they be used? As is the situation with the taper of CS in PMR, a biomarker that legitimately gauges disease activity in GCA would be invaluable. For the present, the interpretation of recurring headache or constitutional symptoms, and of the significance of rises in the erythrocyte sedimentation rate (ESR) and the c-reactive protein (CRP), are exercises in clinical judgment. Whether such increases in symptoms or signs are clinically consequential also bedevils formal studies of the treatment of GCA. For example, does a minor headache or a 10-mm bump in the ESR constitute a bona fide “flare” of GCA, thus to be construed as an inadequacy or even failure of a given treatment? In practice, the slavish adjustment of the CS dose against minor rises in the acute phase reactants, without consideration of the patient’s clinical status, is a recipe for CS-related toxicities. Clinical experience suggests that, on the whole, there is a tendency for overtreatment of GCA with CS, which can lead to more morbidity than the disease itself.
The issue of toxicities due to CS has prompted the search for other therapies for GCA, especially those with ostensible steroid-sparing qualities. The benefits of methotrexate in this regard have proved to be so-so, at best. A randomized controlled trial showed no benefit for adjunctive infliximab, though the evident value of TNF-alpha blockade in the treatment of Takyasu arteritis, the histopathology of which resembles GCA, suggests that this approach could be further investigated.17 And there are recent anecdotes touting favorable effects of IL-6 blockade with tocilizumab.18 Controlled studies will be essential to ascertain the role for the latter in the management of GCA. If tocilizumab—or any other therapy—facilitates shorter courses of CS in the management of GCA, will CS-related toxicities in fact be reduced? Will side effects of adjunctive treatments themselves be problematic? Would any of these treatments actually be used as monotherapy? Could they match the certain worth of CS in the prevention of vision loss, with lesser toxicity?
More Issues: GCA and Large-Vessel Involvement
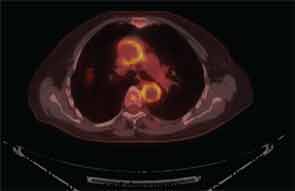
It is now well established that arteries other than the small, above-the-neck, extracranial arteries can be affected by GCA. Findings on FDG PET scanning and arterial ultrasound suggest that subclinical involvement of the large vessels—aorta, great vessels of the neck, and large arteries of the limbs—is common, a major clinical consequence of which is aortic aneurysm, described in 10–18% of reported series (see Figure 3, p. 33).19-22 The questions surrounding the issue of aneurysms in GCA are frustratingly numerous. Who is at risk? What is the relationship to prior CS treatment? What is cost-effective screening? If an aortic aneurysm is identified, how is damage in the arterial wall differentiated from disease activity? What is treatment, and how should it be followed? There are, at present, no good answers, no high levels of evidence to guide decision making. Because appropriate treatment for an asymptomatic aneurysm in patients with GCA is unknown, regular screening with CT of the chest or MRA seems unjustifiable. While we await more robust data, as rheumatologists we must advise patients and their primary physicians as to the potential risk for this issue, and clinical awareness must be sustained, as aneurysms may not manifest for several years after the initial diagnosis of GCA.
A further issue is the discovery of histopathologic aortitis on resection of an apparently routine aneurysm of the ascending aorta. About 5% of these resected aneurysms are inflammatory on histopathologic inspection.23,24 Rare etiologies—e.g., syphilitic aortitis or other infections—are identified in a handful of these cases. Lymphoplasmacytic aortitis, associated with immunoglobulin G-4 disease, is a newly recognized etiology.25 In about one-fifth of cases of inflammatory aortitis, symptoms or signs referable to systemic rheumatic disease—prior PMR or GCA, Takayasu arteritis, relapsing polychondritis, etc.—can be found; the remainder fall under the heading of noninfectious ascending aortitis. Whether this latter subset should be classified along the clinical spectrum of GCA, or understood as a problem sui generis (so-called “isolated aortitis”), is unclear. Work-up and treatment pose challenges similar to the situation of aneurysms occurring in the context of bona fide GCA. Imaging of the arterial tree is warranted, and the acute phase reactants should be measured after the dust has settled from surgery. In the absence of explicit evidence of active inflammation, overtreatment should be avoided, but vigilance for the development of new aneurysms must be maintained, and vascular imaging should be periodically repeated.26
Perhaps further clarification of the immunopathogenesis of GCA will point to new avenues for treatment, as has been the case with bench-to-bedside translations that have culminated in the modern management of rheumatoid disease. The past decade has seen solid progress in unraveling the complexities of T cell biology in GCA, led by Drs. Cornelia Weyand and Jorg Goronzy, professors of medicine at Stanford University in Palo Alto, Calif. Their work has shown that at least two separate lineages of CD4 T cells are functional in GCA, Th1 cells and Th17 cells, the signature cytokines of which are IFN-γ and IL-17, respectively. In GCA, CS suppress TH17 cells, but IFN-γ–producing Th17 cells persist.27 Whether suppression of these prolonged Th1 immune responses would be advantageous in the long-term management of GCA is still conjectural, but it is fundamental investigation of this sort will help to spotlight novel therapeutic targets.
PMR and GCA Considered
Despite the issues surrounding the management of PMR and GCA, we do have a dramatically effective treatment for PMR, in the form of low-dose CS, and an indisputably effective treatment for the prevention of blindness in GCA, the expeditious use of higher doses of CS. Yet PMR often goes unrecognized for weeks or months, and it is striking that the incidence of vision loss in patients with GCA has remained at about 15%, in one series after another, from the U.S. to Europe, over the past two decades.11,28-31 One major explanation for the latter, it seems likely, is that treatment has been delayed because symptoms have not been appropriately recognized.
PMR and GCA are common. The lifetime risk of PMR is only second to RA as a systemic rheumatic disease in U.S. adults, and in adults over the age of 50, the incidence of PMR is probably higher than that of RA.32 GCA is the most common systemic vasculitis by far.33 While awaiting further insights into the pathogenesis of PMR and GCA, better biomarkers for the their diagnosis and follow-up, better ways of managing CS in their treatment, and validation of better therapies, we must be conscientious in the more modest work of educating colleagues in other disciplines about these common systemic rheumatic diseases, so that, in turn, patients may reap the established benefits of timely diagnosis and efficacious treatment.
Dr. Docken is a senior physician at Brigham and Women’s Hospital and assistant professor of medicine at Harvard Medical School, both in Boston.
References
- Weyand CM, Hicok KC, Hunder GG, Goronzy JJ. Disease patterns and tissue cytokines in giant cell arteritis. Arthritis Rheum. 1997;40:19-26.
- Barber HS. Myalgic syndrome with constitutional effects: Polymyalgia rheumatica. Ann Rheum Dis. 1957;16:854-862.
- Healey LA. Long-term follow-up of polymyalgia rheumatica: Evidence for synovitis. Semin Arthritis Rheum. 1984;13:322-328.
- Salvarani C,Cantini F, Boiardi L, Hunder GG. Polymyalgia rheumatica: A disorder of extraarticular synovial structures? J Rheumatol. 1999;26:517-521.
- Dasgupta B, Cimmino MA, Maradit-Kremers H, et al. 2012 Provisional classification criteria for polymyalgia rheumatica: A European League Against Rheumatism/American College of Rheumatology collaborative initiative. Arthritis Rheum. 2012;64:943-954.
- Matteson EL. Clinical guidelines: Unraveling the tautology of polymyalgia rheumatica. Nat Rev Rheumatol. 2010;6:249-250.
- Docken WP. Polymyalgia rheumatica (PMR) can recur years after discontinuation of steroid therapy. Clin Exp Rheumatol. 2009;27:1(Suppl. 52):S25-S27.
- Gonzalez-Gay MA, Vazquez-Rodriguez TR, Lopez-Diaz MJ, et al. Epidemiology of giant cell arteritis and polymyalgia rheumatica. Arthritis Rheum. 2009;61:1454-1461.
- Ball EL, Walsh, SR, Tang TY, Gohil R, Clarke JMF. Role of ultrasonography in the diagnosis of temporal arteritis. Br J Surg. 2010;97:1765-1771.
- Arida A, Kypianou M, Kanakiis M, Sfikakis. The diagnostic value of ultrasoasography-derived edema of the temporal artery in giant cell arteritis: A second meta-analysis. BMC Musculoskel Disord. 2010;11:44.
- Aiello PD, Trautmann JC, McPhee TJ, Kunselman AR, Hunder GG. Visual prognosis in giant cell arteritis. Ophthalmology. 1993;100:550-555.
- Hutchinson J. Diseases of the arteries. Arch Surg (London). 1889-90;1:323.
- Nesher G, Berkun Y, Mates M, Baras M, Rubinow A, Sonnenblick M. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum. 2004;50:1332-1337.
- Lee MS, Smith SD, Galor A, Hoffman GS. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum. 2006;54:3306-3309.
- Narvaez J, Bernad B, Gomez-Vaquero C, et al. Impact of antiplatelet therapy in the development of severe ischemic complications and in the outcome of patients with giant cell arteritis. Clin Exp Rheumatol. 2008;26 (Suppl. 49):S57-S62.
- Salvarani C, Della Bella C, Cimino L, et al. Risk factors for severe cranial ischemic events in an Italian population based cohort of patients with giant cell arteritis. Rheumatology. 2009;48:250-253.
- Schmidt J, Kermani TA, Bacani AK, Crowson CS, Matteson EL, Warrington KJ. Tumor necrosis factor inhibitors in patients with Takayasu arteritis: Experience from a referral center with long-term follow-up. Arthritis Care Res. 2012;64:1079-1083.
- Salvarani C, Magnani L, Catanoso M, et al. Tocilizumab: A novel therapy for patients with large-vessel vasculitis. Rheumatology. 2012;51:151-156.
- Blockmans D, De Ceuninck L, Vanderschueren S, Knockaert D, Mortelmans L, Bobbaers H. Repetitive 18-flourodeoxyglucose positron emission tomography in giant cell arteritis: A prospective study of 35 patients. Arthritis Rheum. 2006;55:131-137.
- Schmidt WA, Seifert A, Gromnica-Ihle E, Krause A, Natusch A. Ultrasound of proximal upper extremity arteries to increase the diagnostic yield in large-vessel giant cell arteritis. Rheumatology. 2008;47:96-101.
- Nuenninghoff DM, Hunder GG, Christianson TJH, McClelland RL, Matteson EL. Mortality of large-artery complication (aortic aneurysm, aortic dissection, and/or large-artery stenosis) in patients with giant cell arteritis: A population-based study over 50 years. Arthritis Rheum. 2003;48:3532-3537.
- Gonzalez-Gay MA, Garcia-Porrua C, Pineiro A, Pego-Reigosa R, Llorca J, Hunder GG. Aortic aneurysm and dissection in patients with biopsy-proven giant cell arteritis from northwestern Spain: A population-based study. Medicine. 2004;83:335-341.
- Liang KP, Chowdhary VR, Michet CJ, et al. Noninfectious ascending aortitis: A case series of 64 patients. J Rheumatol. 2009;36:2290-2297.
- Schmidt J, Sunesen K, Kornum JB, Duhaut P, Thomsen RW. Predictors for pathologically confirmed aortitis after resection of the ascending aorta: A 12-year Danish nationwide population-based cross-sectional study. Arthritis Res Ther. 2011;13:R87.
- Stone JH, Khosroshahi A, Deshpande V, Stone JR. IgG4-related systemic disease accounts for a significant proportion of thoracic lymphoplasmacytic aortitis cases. Arthritis Care Res. 2010;62:316-322.
- Wang H, Smith RN, Spooner AE, et al. Giant cell aortitis of the ascending aorta without signs or symptoms of systemic vasculitis is associated with elevated risk of distal aortic events. Arthritis Rheum. 2012:64;317-319.
- Weyand CM, Younge BR, Goronzy JJ. IFN-γ IL-17–the two faces of T cell pathology in giant cell arteritis. Curr Opin Rheumatol. 2011;23:43-49.
- Font C, Cid MC, Coll-Vinent B, Lopez-Soto A, Grau JM. Clinical features in patients with permanent visual loss due to biopsy proven giant cell arteritis. Br J Rheumatol.1997;36:251-254.
- Gonzalez-Gay MA, Garcia-Porrua C, Llorca J, et al. Visual manifestations of giant cell arteritis. Trends and clinical spectrum in 161 patients. Medicine. 2000;79:283-292.
- Liozon E, Herrmann F, Ly K, et al. Risk factors of visual loss in giant cell (temporal arteritis): A prospective study of 174 patients. Am J Med. 2001;111:211-217.
- Nesher G, Berkun Y, Mates M, et al. Risk factors of cranial ischemic complications in giant cell arteritis. Medicine. 2004;83:114-122.
- Crowson CS, Matteson EL, Myasoedova E, et al. The lifetime risk of adult-onset rheumatoid arthritis and other inflammatory autoimmune rheumatic diseases. Arthritis Rheum. 2011;63:633-639.
- Gonzalez-Gay MA, Garcia-Porrua C. Systemic vasculitis in adults in northwestern Spain,1988-1997. Clinical and epidemiologic aspects. Medicine.1999;78:292-308.