The varicella zoster virus (VZV) can cause persistent inflammation and pathological vascular remodeling diagnosed as giant cell arteritis. VZV vasculopathy can also cause stroke and granulomatous aortitis. The pathologies develop when the virus reactivates from ganglia and spreads transaxonally to arterial adventitia.
The varicella zoster virus (VZV) can cause persistent inflammation and pathological vascular remodeling diagnosed as giant cell arteritis. VZV vasculopathy can also cause stroke and granulomatous aortitis. The pathologies develop when the virus reactivates from ganglia and spreads transaxonally to arterial adventitia.
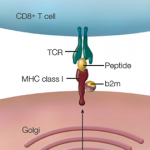
Previous studies have shown that VZV infection causes the upregulation of programmed death ligand 1 (PD-L1) and the downregulation of major histocompatibility complex-1 (MHC-1). Scientists believe that both the virus-induced downregulation of MHC-1 and virus-induced dysregulation of PD-L1 are key to the persistence of the virus, but little is known about how VZV regulates PD-L1 expression.