Identifying Patterns in Vascular Inflammatory Disease
These observations urge further reflection and posing of new questions about the patterns of vascular inflammatory disease.
1) Is nomenclature and classification based on vessel size possibly misleading or an oversimplification of vascular biology? For example, within those illnesses described as “small vessel vasculitides,” not all small vessels share the same vulnerability. Indeed, vasculitis rarely occurs in the microvasculature of the pancreas, adrenal glands, thyroid, tongue, or retina. However, it is relatively common in the skin, lungs, and kidneys.
Among those diseases we call “large vessel vasculitis,” territories of large vessels may differ markedly in their vulnerability to injury. It is more common to find lesions in the proximal than in the distal aorta and in the branch vessels of the arch versus those of the distal aorta.
Our sense of intellectual comfort about understanding the pathogenesis of vasculitis is further compromised by examples of diseases thought to be exclusively small- or medium-sized vessel occasionally affecting the aorta (e.g., systemic lupus erythematosus and Sjögren’s syndrome vasculitis) or, conversely, giant cell arteritis affecting the skin or retinal vessels. What is the basis for this selective vulnerability beyond vessel size?
2) While rare, single-organ vasculitis is a well-known entity that is often cured by surgical resection of the affected component or the entire organ when feasible (e.g., testicle, breast, and gynecologic organ). These examples of presumed limited autoimmunity would theoretically be simpler to study than complex multiorgan vasculitides, and findings may improve our understanding of systemic vasculitis. In what manner are these entities similar to other single-organ immune-mediated diseases (e.g., type I diabetes, hypo- and hyperthyroidism, Addison disease, multiple sclerosis, and primary biliary cirrhosis)?
In many of these examples, abnormal immune responses have been identified to unique single-organ antigens such as insulin, myelin oligodendrocyte glycoprotein, thyrotropin receptor, thyroid peroxidase, and thyroglobulin. Are these immune aberrations to organ-specific proteins proximal, distal, or secondary and inconsequential in pathogenesis? In systemic vasculitides, might shared antigens or homologous antigens in multiple tissues determine multifocal targeting?
3) Is the immune response in each organ unique to that site in some fashion? Are different organs better equipped than others to avoid injury or clear pathogens without resultant harm to the site? Might this in part explain “organ selectivity” which indeed may not be a matter of pathogen tropism?
These questions have begun to generate answers, and the results are extremely exciting. The subsequent discussion will focus on the following issues:
- Early development and modification of vessels during embryogenesis and subsequent life;
- The striking differences that exist between vessels of the same size in different organs, as well as differences that exist even in the proximal and distal territories of the same vessel; and
- Unique immunologic profiles of different vessels and their implied susceptibility or resistance to pathogens.
Embryogenesis
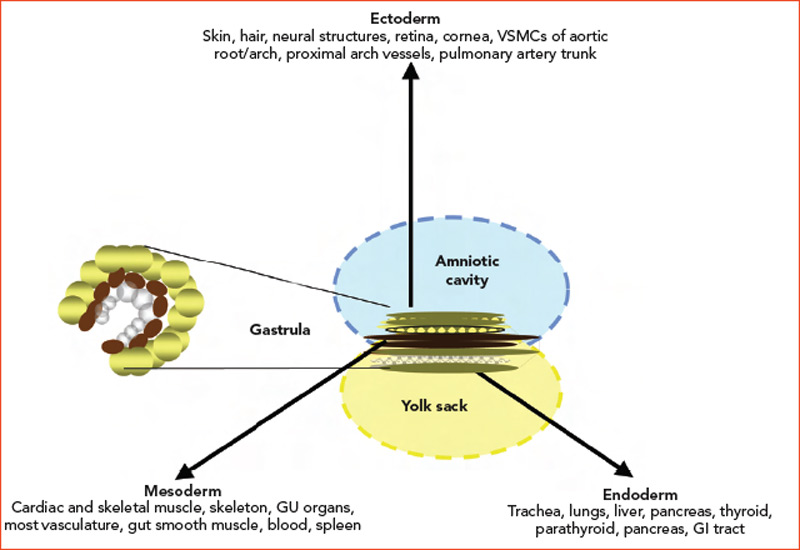
During the third week of human embryonic development, the progenitors of individual organ systems are already present. Undifferentiated cells have become ectoderm, mesoderm, and endoderm. These three layers and their derivatives are represented in Figure 1 (above). The genetic program in nascent organs unfolds in a fashion that is not only unique in regard to the specialized roles of formative parenchyma but also unique to the vasculature and other constituent structures (e.g. nerve, matrix, and lymphatics). The genetic program will change over time, and the products of developing cells may in turn influence the development of neighboring cells. Embryologists speak of these events as “context dependent.” The results may vary depending on context: time and place.
It is critical to note that not all of the vasculature shares a common embryologic source and that a variety of signaling pathways ultimately determine whether early vessel precursors (angioblasts) become veins or arteries. If angioblasts are exposed to vascular endothelial growth factor and subsequently experience activation of membrane-bound Notch receptors (which play a role in development of most vertebrate organs), development moves in an arterial direction. The absence of Notch activation and, alternatively, engagement of COUP-TFII leads to venous development. Ultimately, arteries develop into elastic conducting vessels (aorta and proximal primary branches), and the more distal vessels become muscular resistance conduits.
Diversity within the vascular tree is profound for vessels of all calibers. For example, it would be incorrect to assume that the vascular tube we call the aorta is the same throughout its length. Indeed, it is quite different. It was already noted that the vascular smooth muscle cells (VSMCs) of the root and arch have different embryological origins, and perhaps related to this are known differences in “tube” diameter, wall thickness, elasticity (reduced from proximal to distal), and collagen content (increased proximal to distal). It is also known that proximal and distal aortic VSMCs differ in their ex-vivo response to cytokine stimulation.1
Most relevant to clinicians are whether and how such differences affect disease susceptibility. Aortic arch aneurysms are principally due to cystic medial degeneration, whereas abdominal aortic aneurysms are almost exclusively due to atherosclerosis. This observation has fascinated students of vascular biology, including surgeons and vascular geneticists, who have discovered that approximately 17% of the expressed genome in the proximal versus distal aorta is distinct.2 This is a likely basis for differences in biologic behavior, including responses to both internal and external stimuli. Might these differences in gene expression play a role in aneurysm formation due to aortitis? These aneurysms are far more common in the aortic arch either as an isolated finding or as part of giant cell arteritis or Takayasu arteritis.
Can a similar case for vascular diversity be made in reference to the territories of small vessels? Indeed, it can. In attempting to demonstrate unique qualities of microvascular beds, embryologists and vascular biologists have noted that endothelial cell (EC) gene expression and proteins produced by ECs are as unique as the different organs they perfuse.3,4 In animal studies, transfer of ECs from one organ to another is associated with either EC death or development of abnormal EC characteristics.
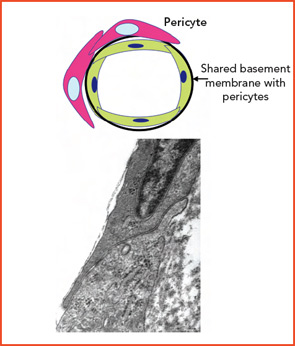
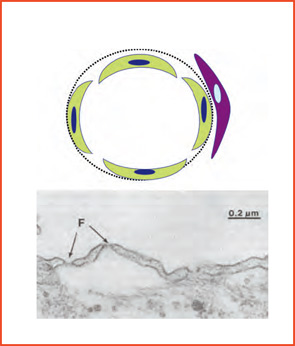
Diversity among small-vessel ECs extends beyond the cells themselves. Microvascular channels within different organs may be supported by surrounding matrix components that differ both qualitatively or quantitatively (e.g., including within vascular basement membranes: collagens, nidogens, laminins, fibronectin, and fibrillin). Adjacent ECs in different organs may function in a profoundly different manner based on physical and physiological relationships with basement membrane and surrounding cells (e.g., pericytes, SMCs, organ parenchymal cells).5,6 (See Figures 2A and 2B, p. 17.)
These relationships form the basis for selectivity of transvascular passage of both cells and chemicals. Where passage is most permissive is within the discontinuous (sinusoidal) endothelium of the liver; it is most selective within the continuous endothelium of the retina and brain. Consistent with the known plasticity of vascular biology, especially during early development, are changes that may occur to adapt to needs such as the early brain containing fenestrated ECs and later the more mature brain having a continuous EC microvascular bed. From a broad range of data that spans macro to microvascular, physical properties to genetic and molecular arrays, it is clear that vascular territories and their component cellular and matrix elements are as unique as the organs they perfuse.
Age-Related Change
The aging process is associated with fragmentation of elastic fibers, depletion of elastica, and an increase in collagen deposition. The most significant changes occur in conjunction with an increase in vessel-wall concentrations of matrix metalloproteinases in muscular/elastic vessels. Coupling of vessel and tissue proteins with reducing sugars leads to production of advanced glycation end products that alter the function of normal proteins and may change their antigenicity. Evidence of progressive EC dysfunction includes a reduced capacity to produce nitric oxide and an increased engagement of pathways of oxidative stress that enhance risk of atherosclerosis and thrombosis. These changes may vary in severity in different sites.8-10
Over our lifespans, we experience innumerable interactions with our environment, acquiring, in some cases, inanimate materials by transcutaneous, airborne, or ingested pathways. Other acquisitions include living materials that were transmitted as early as during gestation (e.g., microchimerisms, or cells from our mothers), or later from others or the environment (e.g., infectious agents, especially herpes viruses). Each may have favorite locations within tissue, including vessels, where they may be of no consequence or could lead to overt infection or changes in tissue antigen expression.
Immunologic Susceptibility
In a seminal study, the laboratory of Cornelia Weyand and Jorg Goronzy extended its prior observations, identifying and characterizing Toll-like receptors (TLRs) in temporal arteries, particularly at the adventitial–medial borders. They have now asked the question of whether or not disease patterns may be related to unique innate immune profiles within specific vascular regions. They compared expression of TLRs 1–9 in the thoracic aorta, carotid, subclavian, temporal, mesenteric, and iliac arteries.11 Each artery was found to possess a unique TLR portfolio. TLRs 2 and 4 were consistently present in all arteries studied; TLRs 7 and 9 were infrequent; and the greatest variability was noted for TLRs 1, 3, 5, 6, and 8. Using their well-known SCID chimera model, they were able to demonstrate that lipopolysaccharide (LPS)-stimulated mice infused with CD 4+ lymphocytes could establish an effective adaptive immune response, whereas controls (unstimulated) did not. If dendritic cells had been depleted by stripping the adventitia from the SCID-embedded vessels, CD4 cells did not home to the vessel. This work established that adventitial–medial dendritic cells are critical to pathogen “sensing” and that the ability of muscular arteries to achieve this goal varies between vessels from different territories.
These observations raise important questions about a fascinating animal model of aortitis initially studied more than 10 years ago.12,13 A usually nonpathogenic herpes virus (γHV68) was provided as a standard inoculum to immunologically intact mice, as well as IFNγ- and IFNγ−receptor(R)-deficient mice. While the intact mice did not develop illness, the IFNγ- and IFNγ−R-deficient mice died from aortic rupture or dissection at approximately eight to 10 weeks. When animals were serially sacrificed and studied from one to six weeks postinoculation, it was apparent that widespread viremia had occurred but that virus could be effectively cleared from the IFNγ- and IFNγ–R-deficient mice from all organs except the aortic root. Thus, despite their lack of either IFNγ- or IFNγ−R, the gene-deficient animals possessed adequate immunological redundancy in other organs and within the distal thoracic and abdominal aorta to clear virus. However, the aortic root was uniquely vulnerable.
Conclusions
Vessels are not merely anatomic conduits that carry blood, gases, nutrients, and waste products. Furthermore, they cannot be considered in simplistic terms based only on vessel size. It is clear that vessels, even of the same caliber from different organs, have important differences in their microanatomy, chemistry, and function. They work as partners in tissue development and have unique barrier and immunologic roles in different sites. Vessels, like all other tissues, also undergo changes in time. They can be sites of topographically selective chemical and other forms of injury or can selectively harbor infectious agents. The injury–repair process that is part of adapting to life’s challenges modifies substrate and is susceptible to errors in response and repair that may include mutations. Although this process plays an important role in the pathogenesis of cancer, it has not been examined within affected vessels in vasculitis. Might mutations provide neo-antigens as one means of stimulating an immune response against vessels, and could this response even be a functionally “normal” attempt to clear a “foreign” protein?
These questions do not diminish the importance of gender effects, immunological senescence, or inheritance in establishing disease predisposition. They do, however, emphasize that vessels are site-variable, complex, specialized organs, with qualities that are probably important in determining disease patterns. We can ill afford to ignore the unique properties of vascular territories and how they change over time if we wish to better understand the pathogenesis of the vasculitides. We should begin to apply lessons from embryology and vascular biology and reach beyond the simplistic notion of vasculitis being “understood” based principally on vessel size.
It is the hope that new molecular and genetic techniques can take advantage of these vessel differences and bring us closer to a more sophisticated understanding of disease etiology and phenotypes.
Dr. Hoffman is professor and Harold C. Schott Chair of Rheumatic and Immunologic Diseases at the Cleveland Clinic Lerner College of Medicine and founder of the Cleveland Clinic Center for Vasculitis Care and Research.
References
- Topouzis S, Majesky MW. Smooth muscle lineage diversity in chick embryo. Devel Biol. 1996; 178:430-445.
- Absi TS, Sundt TM 3rd, Tung WS, et al. Altered patterns of gene expression distinguishing ascending aortic aneurysms from abdominal aortic aneurysms. J Thor Cardiovasc Surg. 2003; 126:344-357.
- Red-Horse K, Crawford Y, Shojaei F, Ferrara N. Endothelium-microenvironment interactions in the developing embryo and in the adult. Developmental Cell. 2007;12:181-194.
- Chi J-T, Chang HY, Haraldsen G, et al. Endothelial cell diversity revealed by global expression profiling. Proc Ntl Acad Sci USA. 2003;100:10623-10628.
- Hallmann R, Horn N, Selg M, Wendler O, Pausch F, Sorokin LM. Expression and function of laminins in embryonic and mature vasculature. Physiology Rev. 2005; 85:979-1000.
- Stan RV. Endothelial stomatal and fenestral diaphragms in normal vessels and angiogenesis. J Cell Mol Med. 2007; 11:621-643.
- Hoffman GS. Disease patterns in vasculitis —still a mystery. Bull NYU Hosp Jt Dis. 2008;224-227.
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle differentiation in development and disease. Physiology Rev. 2004; 84:767-801.
- Najjar SS, Scuteri A, Lakatta EG. Arterial aging. Is it an immutable cardiovascular risk factor? Hypertension. 2005;46:454-462.
- Taddei S, Virdis A, Ghiadoni L, Versari D, Salvetti A. Endothelium, aging, and hypertension. Curr Hyperten Rep. 2006; 8:84-89.
- Pryshchep O, Ma-Krupa W, Younge BR, Goronzy JJ, Weyand CM. Vessel-specific toll-like receptor profiles in human medium and large arteries. Circulation. 2008; 118:1276-1284.
- Weck K, Dal Canto AJ, Gould JD, et al. Murine γ herpesvirus 68 causes severe large-vessel arteritis in mice lacking interferon-γ responsiveness: a new model for virus-induced vascular disease. Nat Med. 1997;3:1346-1353.
- Dal Canto AJ, Swanson PE, O’Guin AK, Speck SH, Virgin HW. IFN-γ action in the media of the great elastic arteries, a novel immunoprivileged site. J Clin Invest. 2001;107:R15-R22.