‘We knew clinically about patients with systemic inflammatory disease & myelodysplasia; we just didn’t realize that there was a unifying process driving these conditions.’ —Dr. Grayson
Kenneth Warrington, MD, professor of medicine and chair of the division of rheumatology at the Mayo Clinic, Rochester, Minn., hailed the “landmark discovery” as a big step toward understanding the root cause of VEXAS and improving therapeutic interventions. “I think where there is more to be discovered is in how we end up seeing this varied clinical phenotype,” Dr. Warrington says, citing potential interactions with other genes or environmental factors as modulating influences. “It’s obviously not just one gene with one disease, but one gene with a more varied clinical syndrome, which I think, in essence, makes it more interesting and probably why it took so long to identify this.”
Multiple Investigative Lines

Dr. Grayson
To discover the disease, Dr. Grayson says the team’s geneticists at the National Human Genome Research Institute (NHGRI) “inverted the script” by initially ignoring the variable clinical symptoms and instead poring through an extensive database of exome sequences collected from the National Institutes of Health’s (NIH’s) Undiagnosed Diseases Program. Starting with a list of target genes, they looked for similarities at the genetic level and found a cluster of three cases that had acquired a methionine-41 mutation in UBA1.
The geneticists, Dr. Grayson says, astutely noticed the patients had two separate sequencing reads in their blood cells: a wild type and a variant UBA1 version. Because the gene is located on the X chromosome, men should have only a single read. Instead of a sequencing error, the researchers realized they were seeing an acquired somatic mutation resulting in mosaicism. “So maybe there will be more diseases to come that are X-linked mutations that are mosaics in men that we just haven’t been looking for,” Dr. Grayson says.
After identifying the first three patients with the UBA1 mutation, the team noticed the patients’ bone marrow biopsy slides all revealed numerous white cellular vacuoles that were likely filled with accumulated protein debris and the byproducts of cellular stress (see Figure 1, above). Collaborating hematopathologists realized they had seen the same clinical feature in other cases, helping add more VEXAS diagnoses to the tally. The team eventually discovered another 15 through the NIH’s Undiagnosed Diseases and Periodic Fever Syndrome databases, and another seven through collaborators in the U.K.
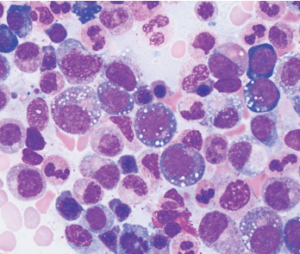
courtesy Katherine Calvo / NIH
Of the 18 patients with available bone marrow biopsies, all had the same vacuoles. Dr. Grayson says they may provide an important new clue for other hematopathologists, while hematologists can now add the UBA1 mutation to a bone marrow-based genetic screen for myelodysplastic syndrome.
Before the new finding, the 25 men had been tentatively diagnosed with a range of inflammatory disorders, such as relapsing polychondritis (RP), polyarteritis nodosa (PAN) and giant cell arteritis. Most were refractory to conventional immunosuppressive agents, developed progressive bone marrow failure and found relief only through escalating doses of glucocorticoids. Of the study’s patient cohort, 40% eventually died. “We knew clinically about patients with systemic inflammatory disease and myelodysplasia; we just didn’t realize that there was a unifying process driving these conditions,” Dr. Grayson says.
Thanks to rapidly growing databases of patient-isolated sequences, more gene-first explorations are likely to yield additional hits. “This is the first disease with a somatic mutation causing late-onset inflammation, but there will likely be others to come,” Dr. Grayson says. For VEXAS, he expects the clinical spectrum will expand when clinicians begin re-investigating more unexplained cases and find the syndrome lurking in other tentative diagnoses. The upshot, he said, is that rheumatologists will need to work closely with geneticists, hematologists and pathologists to resolve late-onset cases that can’t be diagnosed at the bedside.
A big challenge, he and Dr. Warrington agree, is to translate the findings into better treatment options. One major obstacle for treating VEXAS patients, for example, is the associated hematologic disorder that leads to anemia and low blood counts. Such symptoms complicate the administration of rheumatologic drugs that can reduce blood counts as a side effect, further emphasizing the importance of a collaborative approach with hematologists.
As potential therapies, Dr. Grayson says researchers are exploring whether bone marrow transplants might provide one curative option, though the strategy is not without risks. On a longer-term basis, gene editing might allow researchers to harvest the defective myeloid stem cells, edit the mutation and return the corrected copies to the patients’ bone marrow.
“What I’m most excited about is to put this disease out into the world and just see what comes back to us, because we know that we’re underestimating the prevalence of this disease,” Dr. Grayson says. Creating more awareness among rheumatologists and hematologists, he adds, could potentially identify previously undiagnosed cases and bring to light any effective treatments already in use, even in the absence of a clear diagnosis.
“We are all ears to hear what the communal mind brings back to us, because our number one priority is to try to use this information to establish effective treatments for these patients,” he says.
Bryn Nelson, PhD, is a medical journalist based in Seattle.