A dipose tissue has longed been viewed as a harmless tissue in the pathogenesis of chronic inflammatory connective tissue and joint diseases, with fat providing the soft surroundings for damage inflicted by other mechanisms. This dogma, based on the protective role of leptin in metabolic diseases, has long been popular, although it may have hindered thorough basic science research in this field. However, three discoveries have completely changed this point of view and will be the subject of this article.1,2
These advances can be summarized briefly as follows:
In the light of the lively ongoing discussion around the role of osteoimmunology in rheumatic diseases (discussed in “A Duet of Bone and the Immune System” in the July 2011 issue of The Rheumatologist, p. 1), an interesting feature of adiponectin is its potential to stimulate proliferation and receptor activator for nuclear factor κB ligand (RANKL) expression in osteoblasts, whereas osteoprotegerin expression is inhibited.5 Moreover, globular adiponectin strongly inhibits TNF/RANKL-induced osteoclastogenesis as well as osteoclast formation induced by innate immune system arthritis-related Toll-like receptor 4 ligand and RANKL.
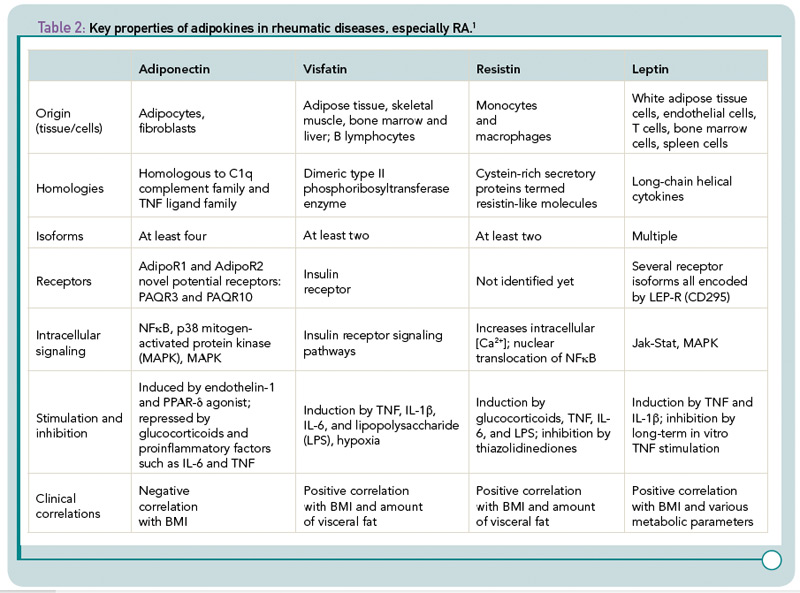
Among other adipokines, resistin has been found in mice as a protein involved in adipocyte differentiation. Human resistin, on the other hand, increases the secretion of proinflammatory cytokines such as TNF, IL-6, and IL‑12, activates endothelial cells and increases endothelin-1 and several adhesion molecules as well as chemokines in these cells.6 Visfatin/pre–B cell colony enhancing factor (PBEF) is also called Nampt because of its nicotinamide phosphoribosyltransferase (NAmPRTase) activity. This molecule is involved in the synthesis of nicotinamide adenine dinucleotide, an essential co-factor of cell metabolism; in addition, it can activate human leukocytes and induces costimulatory molecules on the cell surface. In monocytes, visfatin/PBEF can stimulate the synthesis of proinflammatory cytokines such as IL‑1, TNF, and IL-6, and protects fibroblasts and neutrophils from apoptosis.7
So, how do adipokines influence the course of a chronic rheumatic disease such as rheumatoid arthritis (RA)? Given that levels of adiponectin in synovial fluid and serum are high and are related to joint destruction and long-term disease, these molecules would be expected to have an important role.8 Although articular “fat” is also found around an inflamed joint, the truly important finding on the role of adipokines in pathogenesis of RA related to local expression of adiponectin in joint tissue, including in the lining layer, perivascular area, and in several cells in the sublining (see Figure 1, above); adiponectin expression was also found at sites of cartilage destruction, synovial angiogenesis, and inflammation.
Regarding a role in pathogenesis, adiponectin itself can boost joint-destructive RA synovial fibroblasts to full power by inducing synthesis of proinflammatory cytokines such as IL-6, a variety of chemokines, proangiogenic factors such as vascular endothelial growth factor, and matrix-degrading proteins including collagenases.9,10 This pathway appears to be independent of the known TNF routes because TNF and IL-1 could not be induced by adiponectin in RA synovial fibroblasts. The pluripotency of adiponectin was further illustrated by a similar effect on the rest of the “damaging orchestra of cells” in the rheumatoid joint. These cells, which play both loud and out of tune, include endothelial cells, chondrocytes, and lymphocyte subpopulations.

Evolving Picture of Adipokine Action
Things are never as simple or easy as they first appear, however. As shown in subsequent studies, it has become obvious that human adiponectin consists of different isoforms and that glycosylation and oligomerization, which differ between species, are very important for the resulting functional effects of adiponectin.10,11 Because most experiments and animal models are performed using a “wild-type” mix containing different amounts of the respective isoforms, it is not yet possible to fully determine which isoforms are dominant in RA synovial tissue at sites of cartilage degradation and inflammation.
Along this line, inhibiting the synthesis of adipnectin or its downstream effects would appear to be a promising therapeutic goal.5 The key question is, Does it work? The results on this issue are puzzling. Standard treatment with methotrexate appears to increase adiponectin levels in humans and application of a cyclooxygenase-2 inhibitor increased adiponectin in an arthritis animal model. Although adiponectin resembles TNF in its tertiary structure, the true effect of TNF inhibitors on adiponectin expression also still needs to be determined.12,13 Interestingly, experimental treatment with anti-TNF antibodies can downregulate adiponectin-dependent stimulation of synovial fibroblasts significantly.9 This finding also raises the question of whether TNF inhibitors are truly specific for TNF (which, if they are not, might even be an advantage for the patients).
Resistin is another powerful adipokine that is synthesized in the lining layer by macrophages, B cells, and plasma cells, all strongly operative in rheumatoid pathophysiology.6,14 Resistin concentrations in synovial fluid have been found to be higher than in plasma and correlated positively with levels of several inflammation markers including C-reactive protein (CRP), erythrocyte sedimentation rate, IL-1Ra, and TNF-α. Resistin, in turn, is inducible by TNF-α, IL-6, and IL‑1β in peripheral blood mononuclear cells (PBMCs), resulting in a positive feedback mechanism of inflammation. These effects can be reduced by blocking the key transcriptional element NF-κB, leading to the idea that resistin-induced inflammation is NFκB dependent. However, the true potential of this adipokine in the pathogenesis of arthritis is illustrated nicely by the effects of direct injection in joints of arthritis-susceptible mice. In this case, inflammation and joint destruction started immediately and in a rapidly progressive manner. Of note, anti-TNF treatment of RA patients seems to reduce resistin serum levels—as well as CRP—significantly.12
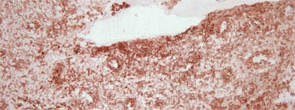
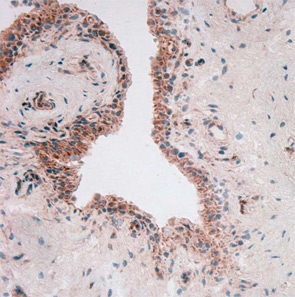
To throw adipose tissue–adipokine restriction dogma further overboard, another adipokine had to be introduced into this challenging scientific game: visfatin/PBEF. Lymphocyte aggregates and vascular endothelial cells predominantly synthesize visfatin in rheumatoid synovium (see Figure 2). Visfatin activates human leukocytes, induces co-stimulatory molecules on the cell surface of monocytes, and induces proinflammatory cytokines such as IL-1, TNF, and IL-6 in these cells; it also protects fibroblasts and neutrophils from apoptosis.7
Protective Adipokines
One question is frequently asked: Where are the “protective” adipokines? Similar to the proinflammatory cytokines, there should be at least one. So let’s therefore revisit the effects of “protective” leptin. The basis for this positioning is the observation that leptin-deficient ob/ob mice developed a less severe arthritis in comparison with wild-type mice. In humans, levels of leptin are positively correlated with the overall body fat content, but a true correlation between inflammation and leptin plasma concentrations is still under discussion.
With respect to the role of leptin in RA, leptin serum levels are higher in active disease. Furthermore, the differences in leptin levels between plasma and synovial fluid were greater in nonerosive than in erosive arthritis, supporting the idea of a joint-protective role. However, studies have indicated that known antirheumatic treatments, such as anti-TNF therapy of RA patients, do not influence concentrations of leptin in serum. To elucidate fully the role of leptin in clinical immunology and rheumatology, compartment-related analyses still need to be performed.
The differences in leptin levels between plasma and synovial fluid were greater in nonerosive than in erosive arthritis, supporting the idea of a joint-protective role.
Usually, the effect of proinflammatory signalling molecules does not differ extensively in the various rheumatic diseases. Does this other dogma also hold true for adipokines?
Given that the differential diagnosis of a mild RA versus active osteoarthritis (OA) can be difficult, the identification of a disease-specific adipokine in this setting would be of value. Although studies addressing the role of adipokines in OA are limited, increased adiponectin serum levels appear to be associated with erosive OA, and serum levels have been found to be increased in comparison to healthy individuals. Moreover, visfatin was able to upregulate proinflammatory prostaglandin E2 release in murine arthritis, most likely via activation of the canonical IL-1-chondrocyte-matrix-metalloproteinase pathway.
A similar issue on the role of adipokines in disease exists for the spondyloarthropathies, in which inflammation parameters are generally low and not very useful for (differential) diagnosis. Here, serum levels of resistin in ankylosing spondylitis (AS) patients were found to be low, but nevertheless greater than those in patients with OA. In contrast to the findings with resistin, studies on AS patients showed lower leptin levels compared with controls even after adjustment for body fat mass. These findings suggest a lack of the assumed protective leptin effect. “Fatty life” is complicated. However, in another study, leptin exerted proinflammatory effects on PBMCs from patients with AS, and stimulation of PBMCs from AS patients with exogenous leptin significantly increased IL-6 and TNF production in a dose-dependent manner.15 These findings point to a more direct role of leptin in driving inflammation. (See Figure 3)
Adipokines in Connective Tissue Diseases and Vasculitides
As the dedicated reader might expect, the pathophysiology of connective tissue diseases and vasculitides, especially of systemic lupus erythematosus (SLE), includes also a potential role of adiponectin. In SLE patients with inflammatory glomerulonephritis, the adiponectin isoforms appear to exert different roles, as full-length adiponectin, containing all isoforms except the globular adiponectin–induced IL-8 and monocyte chemotactic protein 1 in microvascular endothelial cells and monocytes. Similarly, studies have indicated that resistin is related to systemic inflammation in SLE. In contrast, the role of leptin in SLE is linked to hypoandrogenicity, reflected by a negative correlation to androstenedione levels.
Less information is available on the role of adipokines in other connective tissue diseases such as systemic sclerosis (SSc). Serum levels of leptin seem to be decreased in patients with SSc, but further details, especially the role of adipokines in fibrosis, still need to be elucidated.16
Research in vasculitis has only addressed leptin thus far. In studies on antineutrophil cytoplasmic antibody-associated vasculitis, leptin levels were found to be significantly lower than those in healthy controls with or without body mass index (BMI) correction, and leptin levels were correlated negatively with disease activity.17 In contrast, in Behçet’s disease, leptin concentrations were found to be increased during active disease in comparison to inactive periods in this study.18 Along this line, resistin and IL-6 serum levels were significantly higher in patients with Behçet’s disease in comparison to healthy controls.
Conclusion
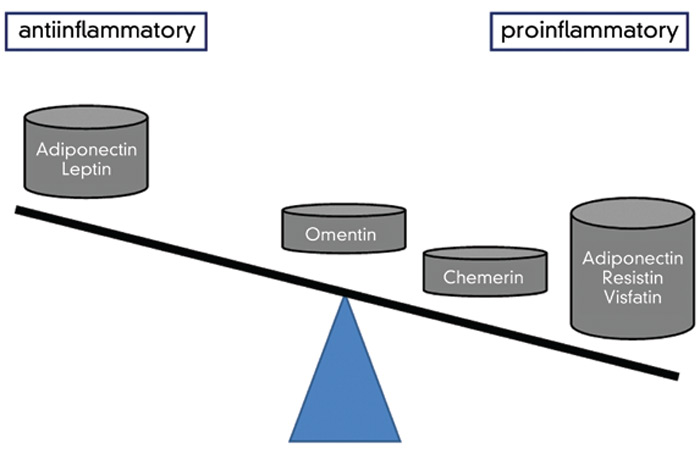
Taken together, studies on the pluripotent effects of adipokines in chronic inflammatory rheumatic diseases, as well as their role in matrix remodelling, are one of the most interesting current topics in rheumatology and other subspecialties involved in the care of patients with chronic inflammatory diseases.5,8,13 While the role of the adipokines is complex, future studies may lead to novel therapeutic strategies or provide explanations for the benefits of current treatments as well as, importantly, the mechanisms for resistance to antirheumatic drugs. This future will also include the detailed evaluation of other members of the growing family of adipokines, (e.g. omentin, chemerin, cartonectin, and many others), which will also stimulate the cross-fertilization between various clinical disciplines (see Table 3).19,20 While fat may seem to be an annoying tissue to patients, as studies of adipokines show, it can be a source of important factors that may have an impact on the pathogenesis of arthritis as well as many other diseases. the rheumatologist

Dr. Müller-Ladner is chair and Drs. Frommer and Neumann are professors of internal medicine and rheumatology at Justus-Liebig-University Giessen in Bad Nauheim, Germany.
References
- Neumann E, Frommer K, Vasile M, Müller-Ladner U. Adipocytokines as driving forces in rheumatic diseases. Arthritis Rheum. 2011;63:1159-1169.
- Schäffler A, Schölmerich J. Innate immunity and adipose tissue biology. Trends Immunol. 2010;31:228-235.
- Rajala MW, Scherer PE. Minireview: The adipocyte—at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. 2003;144:3765-3773.
- Palmer G, Gabay C. A role for leptin in rheumatic diseases? Ann Rheum Dis. 2003;62:913-915.
- Luo XH, Guo LJ, Xie H, et al. Adiponectin stimulates RANKL and inhibits OPG expression in human osteoblasts through the MAPK signaling pathway. J Bone Miner Res. 2006;21:1648-1656.
- Bokarewa M, Nagaev I, Dahlberg L, Smith U, Tarkowski A. Resistin, an adipokine with potent proinflammatory properties. J Immunol. 2005;174:5789-5795.
- Brentano F, Schorr O, Ospelt C, et al. Pre-B cell colony-enhancing factor/visfatin, a new marker of inflammation in rheumatoid arthritis with proinflammatory and matrix-degrading activities. Arthritis Rheum. 2007;56:2829-2839.
- Giles JT, Allison M, Bingham CO, 3rd, Scott WM, Jr., Bathon JM. Adiponectin is a mediator of the inverse association of adiposity with radiographic damage in rheumatoid arthritis. Arthritis Rheum. 2009;61:1248-1256.
- Ehling A, Schäffler A, Herfarth H, et al. The potential of adiponectin in driving arthritis. J Immunol. 2006;176:4468-4478.
- Frommer KW, Zimmermann B, Meier FM, et al. Adiponectin-mediated changes in effector cells involved in the pathophysiology of rheumatoid arthritis. Arthritis Rheum. 2010;62:2886-2899.
- Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288-1295.
- Gonzalez-Gay MA, Garcia-Unzueta MT, Gonzalez-Juanatey C, et al. Anti-TNF-alpha therapy modulates resistin in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2008;26:311-316.
- Popa C, Netea MG, de Graaf J, et al. Circulating leptin and adiponectin concentrations during tumor necrosis factor blockade in patients with active rheumatoid arthritis. J Rheumatol. 2009;36:724-730.
- Moschen AR, Kaser A, Enrich B, et al. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol. 2007;178:1748-1758.
- Park MC, Chung SJ, Park YB, Lee SK. Pro-inflammatory effect of leptin on peripheral blood mononuclear cells of patients with ankylosing spondylitis. Joint Bone Spine. 2009;76:170-175.
- Kotulska A, Kucharz EJ, Brzezinska-Wcislo L, Wadas U. A decreased serum leptin level in patients with systemic sclerosis. Clin Rheumatol. 2001;20:300-302.
- Kumpers P, Horn R, Brabant G, et al. Serum leptin and ghrelin correlate with disease activity in ANCA-associated vasculitis. Rheumatology. 2008;47:484-487.
- Yalcindag FN, Yalcindag A, Batioglu F, Caglayan O, Kisa U, Ozdemir O. Evaluation of serum resistin levels in patients with ocular and non-ocular Behçet’s disease. Can J Ophthalmol. 2008;43:473-475.
- Catalan, V. et al. Increased adipose tissue expression of lipocalin-2 in obesity is related to inflammation and matrix metalloproteinase-2 and metalloproteinase-9 activities in humans. J Mol Med. 2009;87:803-813.
- Sommer G, Weise S, Kralisch S, et al. Lipocalin-2 is induced by interleukin-1beta in murine adipocytes in vitro. J Cell Biochem. 2009;106:103-108.