Most healthcare professionals do not frequently encounter, or are even actively aware of, amyloid protein–related disorders beyond the most commonly known form, Alzheimer’s disease, which is caused by the aggregation of beta-amyloid proteins in the brain. However, for rheumatologist Jonathan Kay, MD, professor of medicine at the University of Massachusetts Medical School in Boston, his awareness of, and subsequent focus on the wider world of amyloidosis stemmed from a seemingly unrelated patient referral.
Amyloidosis Defined
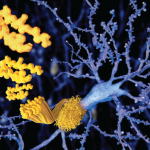
In essence, amyloidosis is a disease caused by protein misfolding. Once expressed and then misfolded, these proteins infiltrate, aggregate, and then form insoluble fibrils that interfere with the normal function of a number of vital organs.
There are at least 18 endogenous human proteins that can be involved in the pathogenesis of distinct types of amyloidosis, each of which forms fibrils in combination with various members of an array of glycosaminoglycans. However, most types of amyloidosis share a specific component—serum amyloid P protein—a critical bit of information when considering imaging modalities for amyloidosis, as discussed below.
Once a nucleus of misfolded proteins forms, it serves as a template for further aggregation, thereby driving the progression of a given amyloidosis disease state.2 The organs/systems involved may include the brain, heart, kidneys, gastrointestinal tract, liver, spleen, peripheral nervous system, and musculoskeletal system.
Types of amyloidosis are grouped as localized, relative to a specific organ (e.g., Alzheimer’s), or systemic. Systemic forms of amyloidosis are further delineated by cause. For primary (AL) amyloidosis, the most common form of the disease, the subunit protein is the immunoglobulin light chain produced in bone marrow. Secondary, or acquired, (AA) amyloidosis typically results from chronic infection or inflammation. This form is rarely seen in the U.S., probably because of the availability of effective antiinflammatory and antibiotic treatments. The amyloidosis that developed in dialysis patients arose because of increased production and inadequate clearance of the subunit protein, β2-microglobulin (Aβ2M). Amyloidosis may also be hereditary in origin, resulting from mutations of transthyretin (ATTR) and other subunit proteins.
AL amyloidosis is generally seen in the elderly and rarely occurs in patients under the age of 40.3 Its occurrence is skewed toward males at a nearly 3:1 ratio, as compared to females. Overall, however, the incidence of amyloidosis is relatively low—only about 2,200 new cases are diagnosed in the U.S. annually. That said, the mortality of this disease is significant; without treatment, the median survival is 13.4 months; survival at 5 years is 7%; and 10-year survival rate is a dismal 1%.4
Of patients with AL amyloidosis, those with peripheral neuropathic involvement fare the best and those with cardiac involvement the worst. “The typical cause of death in a patient with systemic AL amyloidosis is congestive heart failure due to massive infiltration of the heart with amyloid,” said Dr. Kay.
Clinical Features
The clinical features of amyloidosis range from vague to striking. “Patients may present with relatively nonspecific symptoms,” Dr. Kay said. “Weakness, fatigue, unexplained weight loss.”
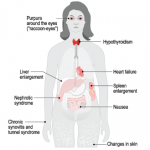
The possibility of fibromyalgia syndrome might first come to mind. However, if present, other obvious clinical features of the disease should prompt a more detailed patient evaluation. These may include waxy papules on the skin surface due to amyloid deposits or nonthrombocytopenic purpura, explained by amyloid deposits binding to clotting factor X, which results in hemorrhages in the skin.
Other features to look for include the otherwise unexplained occurrence of:
- Musculoskeletal
- Noninflammatory arthropathy (“shoulder pads” related to AL, Aβ2M)
- Subcutaneous nodules
- Carpal tunnel syndrome
- Bone cysts
- Destructive spondylarthropathy
- Neurologic
- Sensory neuropathy
- Autonomic neuropathy
- Carpal tunnel syndrome
- Renal
- Proteinuria and/or nephrotic syndrome
- Cardiac
- Congestive heart failure
- Restrictive cardiomyopathy
- Arrhythmias
- Angina
- Pulmonary
- Solitary nodules
- Interstitial infiltration
- Gastrointestinal
- Macroglossia
- Malabsorption
- Diarrhea
- Obstruction
- Ulceration
- Hemorrhage
- Hepatomegaly
- Lymphoid
- Splenomegaly
- Lymphadenopathy
Regarding arthropathy, “In the 1980s, if you walked into a dialysis unit and looked only at the patients’ shoulders … they resembled the hypertrophied deltoid muscles of body builders,” commented Dr. Kay. This was caused by amyloid infiltration of the rotator cuff and surrounding tissues.
(For a more comprehensive review of amyloidosis clinical features, see: Gertz et al. Mayo Clin Proc. 1999;74:490-494.)
Diagnosing Amyloidosis
If one or more clinical features are present, a screening biopsy should be performed and stained with the diazo dye, Congo red. Source tissue for biopsy may include bone marrow, rectal tissue, labial salivary gland, or abdominal fat pad aspiration. Dr. Kay described the process of taking an abdominal fat pad biopsy:
Using a 20-cc syringe with a wide-bore, 14- or 16-gauge needle, first draw sterile saline into the syringe. “This allows you to inject, and withdraw [a volume] repeatedly in order to disrupt the abdominal fat globules,” he said. Then aspirate firmly to draw fat globules into the syringe, and deposit the globules on a glass slide. “Place a second slide on top of that,” he explained, “and squeeze to flatten the fat globules. Allow the preparation to air dry and then send it to the pathology department for fixation and Congo red staining.”
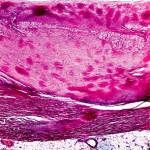
When stained with Congo red and observed under polarized light, amyloid deposits display an apple-green birefringence, a phenomenon of double refraction of light (see Figure 1). Abdominal fat pad aspiration has a positive predictive value of 98%, and a negative predictive value of 76% for diagnosing AL amyloidosis.5
For AL amyloidosis, serum and urine immunofixation electrophoresis will detect a monoclonal light chain in roughly 90% of patients.
If both screening biopsy and serum and urine immunofixation electrophoresis yield negative results, consider that the patient might have a hereditary form of amyloidosis (ATTR). This diagnosis can be confirmed either with isoelectric focusing to detect mutant transthyretin protein or restriction fragment length polymorphism (RFLP) analysis to detect a mutant gene.
If both techniques are negative, rarer forms of amyloidosis should be considered.

Imaging
Imaging modalities for amyloidosis include standard radiographs, MRI, ultrasound, and scintigraphic imaging.
Regarding standard radiography as it pertains to Aβ2M amyloidosis patients, Dr. Kay noted that, “in the axial skeleton, these patients may develop a destructive spondyloarthropathy which looks very much like septic diskitis.” Unless the radiologist is provided with a good history on the patient, or she or he likely will diagnose diskitis and recommend hospitalization with a course of antibiotics for your patient.
The precedent for using ultrasound imaging in AL amyloidosis was set in the late 1980s with echocardiography, used both to gauge disease impact on ejection fraction and to detect cardiac amyloid deposits. Prompted by this, Dr. Kay and colleagues developed criteria by which musculoskeletal ultrasound could be used to diagnose Aβ2M amyloidosis in dialysis patients.6
Scintography using radio-labeled amyloid P component, a ubiquitous constituent of all amyloid deposits, has been used to determine systemic involvement in amyloidosis, however, as Dr. Kay pointed out, this application is investigational, “and fraught with all the potential risks of using a biological product.”
Treatment
No medications are currently approved to treat patients with amyloidosis. Current strategies for disease management include supportive care, reduction of the amyloid precursor protein pool, stabilization of the native structure of precursor protein, and interference with constituents of amyloidosis fibrils.
In many cases, “one must rely on one’s internal medicine background to provide supportive care for these patients, such as when treating neuropathic pain or hypotension due to peripheral, or autonomic, neuropathy,” he said.
Preventative treatment in this setting is primarily directed at AL amyloidosis and, in general, employs regimens historically used to treat multiple myeloma. The goal of treatment is to reduce the amyloid precursor protein pool using:
- Melphalan + predisone ± colchicine
- Melphalan + high-dose dexamethasone
- Melphalan + dexamethasone + either thalidomide, lenalidomide, or bortezomib
- Clyclophosphamide + dexamethasone + lenalidomide
- Syngeneic or allogeneic bone marrow transplant
- Dose-intensive melphalan with autologous stem cell transplantation7
Commenting on this last approach, Dr. Kay said, “regardless of the potential and actual complications, dose-intensive melphalan with autologous stem cell transplantation offered a sea change in treating this devastating disease.” With treatment, estimated overall survival at five years is 47%.7 “That is a dramatic difference, as compared to the era before effective treatment was available,” he said.
For AA amyloidosis, treatment options include chlorambucil, tumor necrosis factor–α inhibitors (they reduce proteinuria), colchicine, and eprodisate, although the last is investigational—eprodisate is a member of a new class of agents that interferes with interactions between fibril proteins and glycosaminoglycans.
For ATTR, treatment options include liver transplant (the liver being the site of the mutant transthyretin expression) and tafamidis. Tafamidis binds to transthyretin, preventing amyloidogenic aggregation. “Use of this drug resulted in a slower rate of worsening neurologic symptoms,” said Dr. Kay. Tafamidis has been approved for use in Europe, but is not yet approved by the U.S. Food and Drug Administration.8
For Aβ2M, treatment may include renal transplantation, which causes a reduction in symptoms due to increased Aβ2M clearance.
The key element of any treatment approach, said Dr. Kay, is timing. “These patients must be treated as early as possible in the disease course to minimize organ involvement.” It is hoped that with greater awareness of the clinical features and diagnosis of amyloidosis, improvements in therapy will finally make significant headway in managing this deadly disease.
Neil Canavan is a journalist based in New York.
References
- Bardin T, Zingraff J, Kuntz D, Drüeke T. Dialysis-related amyloidosis. Nephrol Dial Transplant. 1986;1:151-154.
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583-596.
- Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79:1817-1822.
- Kyle RA, Gertz MA. Primary systemic amyloidosis: Clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45-59.
- Gertz MA, Li CY, Shirahama T, Kyle RA. Utility of subcutaneous fat aspiration for the diagnosis of systemic amyloidosis (immunoglobulin light chain). Arch Intern Med. 1988;148:929-933.
- Kay J, Benson CB, Lester S, et al. Utility of high-resolution ultrasound for the diagnosis of dialysis-related amyloidosis. Arthritis Rheum. 1992;35:926-932.
- Skinner M, Sanchorawala V, Seldin DC, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: An 8-year study. Ann Intern Med. 2004;140:85-93.
- Said G, Grippon S, Kirkpatrick P. Tafamidis. Nat Rev Drug Discov. 2012 ;11:185-186.