The economic implications of biologic therapies are staggering. In 2009, 75% of small molecules (pills and capsules) dispensed were in generic form. By contrast, biologic medications account for 43% of the Medicare Part B budget! When will generic biologic disease-modifying antirheumatic drugs (DMARDs) become commercially available?
Biologics Go Global
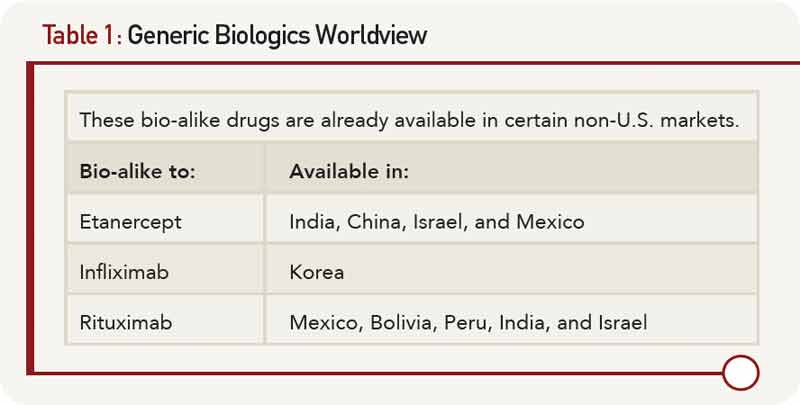
Although patent expiration in the U.S. for several biologic therapies may be several years away (etanercept’s patent expires in 2026, infliximab’s and rituximab’s in 2018, adalimumab’s in 2017), biosimilars for these products are currently under development. In countries where the regulatory environments are much less stringent than in the U.S. or Europe, there already exist “bio-alike” products. In these countries (listed in Table 1), the cost of development has been significantly reduced, in part because randomized controlled trials to formally demonstrate equivalency have not been required.
Though these products have not undergone the stringent regulatory reviews required in the U.S., E.U., Canada, or Australia to demonstrate biosimilarity, economic pressures have propelled their development in those countries listed in Table 1.
Accordingly, in 2009 Congress passed legislation to address the process of bringing biosimilars to the U.S. market as part of healthcare reform. Termed the Biologics Price Competition and Innovative Act, this legislation defines the requirements for a biosimilar compared to the original “innovator” product as follows:
- Must be “highly similar” to the original product;
- Must involve the same mechanism of action;
- Must have no clinically meaningful differences in potency, purity, or toxicity;
- Must be expected to produce the same clinical results; and
- Must utilize the same route of administration, dose, and strength.
A biosimilar may be determined to be sufficiently similar (i.e., “highly similar”) to the innovator product when it can be interchangeable with the original, such that no significant differences would be expected to occur if the patient were to be switched back and forth between the innovator drug and the biosimilar.
Currently Available Biosimilars—And Their Complications
Biosimilars for human growth factors and hormones have already been developed and brought to market in the E.U. These include epoietin, insulin, filgrastim, and somatatropin. These are complex molecules; for example, somatotropin has a molecular weight that is 100 times greater than aspirin. The advent of a less costly biosimilar, filgrastim (cf. Neupogen), resulted in a dramatic increase in the use of this agent in the U.K. On the other hand, the development of the erythropoietin biosimilar has faced many challenges. Seemingly small changes in manufacturing have led to dramatic consequences. For example, the tungsten-containing component of the rubber plunger that was used in the prefilled syringe resulted in unfolding of the protein. This change in structure led to the formation of antibodies to the biosimilar that cross-reacted with native erythropoietin. As a result, between 1998 and 2004, there were 175 cases of pure red-cell aplasia in patients taking the drug. The newly approved erythropoietin biosimilar (Omontys) was recalled due to patients developing allergic reactions, some of which were fatal.
The monoclonal antibodies (mAbs) and receptor fusion proteins display yet another order of magnitude in molecular weight and complexity compared to growth factors and hormones. One could easily imagine how a slight change in protein folding or glycosylation could produce a change in function or immunogenicity compared to the innovator biologic. In fact, there is historical precedent for this phenomenon. Lenercept, a p55 TNF receptor (TNF-RI) fusion protein, was developed at about the same time as etanecept (p75 TNF-RII). However, it never reached the marketplace due to a number of manufacturing problems that resulted in different glycosylation patterns and varied clinical efficacy.
Clinical trials of the biosimilar in humans will need to demonstrate that it is indeed “biosimilar.”
Biosimilars for the U.S. Market
Assuming that a biosimilar manufacturer can derive the exact amino sequence of a mAb or fusion protein receptor (apparently possible, but incomprehensible to the nonscientist), the very manufacturing process is critical. In fact, when the Food and Drug Administration (FDA) approves an innovator biologic DMARD, the approval applies to this precise production process. Any subsequent change in the manufacturing process must be approved by the FDA, which has the power to require additional studies, including clinical trials, to verify the safety and efficacy of the new process.
FDA guidance documents based on the Biologics Price Competition and Innovative Act specifically stipulate the requirements for successful registration of a biosimilar in the U.S. At least two separate clinical studies of immunogenicity are required, pre- and postapproval. At least one study will be required for pharmacokinetics and pharmacodynamics, as well as additional analytical studies with details to be specified, based on the product. Clinical trials of the biosimilar in humans will need to demonstrate that it is indeed “biosimilar,” e.g., “equivalent” in terms of efficacy, pharmacokinetics and pharmacodynamics, immunogenicity, and with a safety profile very similar to the innovator product. Finally, extrapolation from one clinical indication to other indications that have been approved for the innovator will be decided on a case-by-case basis.
Despite the daunting challenges, manufacturers believe that, even with these stringent requirements (which are as equally demanding in Europe as in the U.S.), the costs of developing and bringing to market a biosimilar would be far less than the cost of developing a novel agent. The first test of this hypothesis is about to start. In June 2013, the European Medicines Agency approved the marketing of an infliximab biosimilar, Remsima. This drug is manufactured in South Korea by Celltrion. It is expected to reach the U.S. market following the expiration of the infliximab patent.
Dr. Strand is clinical professor, adjunct, in the division of immunology/rheumatology at Stanford University in Stanford, Calif. She is an independent biopharmaceutical consultant in clinical development and has consulted for: Abbvie, Amgen Corporation, AstraZeneca, BiogenIdec, BMS, Genentech/Roche, GlaxoSmithKline, Janssen, Lilly, Novartis, Pfizer, Regeneron, Sanofi, and UCB. Dr. Brasington is professor and head of the fellowship program at Washington University in St. Louis.