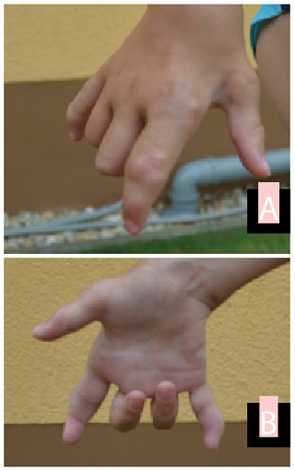
On examination, he presented with significant limitation of motion of fingers II through V of both hands with thickening of the flexor tendon sheaths. He also had swelling of the ankles, knees, wrists and elbows, with significant pain and limitation of motion. Ultrasound at the time of presentation showed only mild effusions in the elbows, with no synovial thickening, but tenosynovitis of the flexor tendon sheaths. No subcutaneous nodules were noted.
Laboratory investigations showed normal CBC, negative CRP and ESR, and normal values for renal and liver function tests. Serum immunoglobulin levels for IgG, IgA and IgM were within normal range. Rheumatoid factor and antinuclear antibodies were not present, and HLA-B27 was negative.
He was tentatively diagnosed with juvenile idiopathic arthritis (with a subtype according to ILAR criteria of rheumatoid factor-negative polyarthritis) and was treated with indomethacin (2.7 mg/kg/day, divided three times per day), subcutaneous methotrexate (12.5 mg/m2 BSA/week) and several courses of methylprednisolone, which led to only temporary improvement.
After three months, he was also started on etanercept (0.89 mg/kg/week), which led to moderate improvement in pain and general mobility, but not in the range of motion. Starting at age 3 years, he started to develop nodules along the flexor tendons of the hand, as well as the back of the hand and on the dorsal side of the toes.
At that time, the increasing nodules, hoarseness and resistance to antiinflammatory treatment with progressing contractures led to the suspicion of Farber disease. Fibroblast cultures showed highly elevated levels of undegraded ceramide compared with normal controls. A genetic analysis of the ASAH1 gene revealed a homozygous mutation in exon 10 (c.770T>c; p.Leu257Pro). Genetic analysis of the parents confirmed the presence of heterozygous mutations of the same type in both parents.
After the diagnosis was confirmed, treatment with methotrexate and etanercept was discontinued. A course of chloroquine did not lead to significant improvement. Due to worsening joint pains, etanercept was reinstituted at approximately age 5 years, leading to significant improvement in reported pain and mild improvement of arthritis. However, limitations of motion in all large joints, as well as induration and contracture of flexor tendons in the hands progressed despite intensive physiotherapy. A course of injections of dexamethasone in the flexor tendons led to only transient improvement.
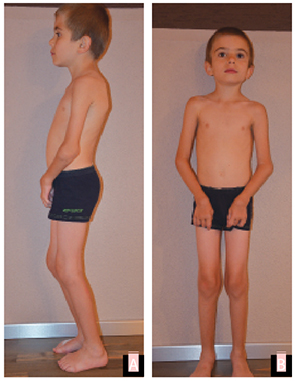
Stem cell transplantation was discussed with the parents. However, due to both religious reservations and concerns about the safety of the procedure, no decision was reached. At last follow-up at age 8 years, the patient continues to be mostly pain-free on etanercept (0.84 mg/kg, divided twice weekly), with significant and ongoing limitation of motion, but able to walk and play nonetheless (see Fig. 1 and Fig. 2).
Discussion
Farber disease is an exceptionally rare disease, with 78 patients described worldwide in 2008.1,4 The gene defect was first described in 1972. To date, about 20 mutations in the gene for acid ceramidase (ASAH1) have been described.5 The Leu257Pro mutation found in this patient has not previously been described in children with Farber’s disease. It does not appear to be associated with neurological manifestations, but significant phenotypic variations have been reported for ASAH1 mutations.6
Farber disease usually presents in patients within the first year of life. A review of 31 patients published in the medical literature shows the median time for onset of first symptoms as 4 months old.7-29 Only two patients were reported to have presented as late as 20 months.3,16 Our patient’s presentation with hoarseness of voice at age 18 months certainly represents the upper end of the spectrum of age at first presentation. It’s important to note that our patient also developed arthritis-like symptoms at 30 months of age, which is later than most patients with Farber disease. It is not surprising, then, that he was initially diagnosed with juvenile idiopathic arthritis.

The similarity between JIA and Farber’s disease has been noted previously. Farber disease can be divided into seven types, of which only the first three comprise more than seven patients reported so far.4,30 According to this classification, this patient would belong to Type 3, which is often regarded as a mild form of the disease. Patients still suffer from the classical triad of symptoms, but they experience slight or no neurological disease and may live well into adulthood. Mild cases of Farber disease are especially susceptible to misdiagnosis due to the under-representation of mild Farber disease cases in the medical literature. However, the nodules and accompanying hoarseness, as well as the early onset and unrelenting progress of the disease provide important clinical clues. A phenotypic comparison of juvenile idiopathic arthritis and Farber disease is shown in Table 1.
With enzyme replacement therapy in preclinical development, current treatment options in Farber disease are limited. The patient has benefited from intensive physical therapy, but still shows unrelenting, progressive disease with increasing contractures and functional impairment. It’s interesting to observe that anti-tumor necrosis factor medication can be effective in alleviating joint pain in this disease, despite its inability to halt its progression.
The only curative treatment for Farber disease available at this time is stem cell transplantation. In cases without neurological involvement, this treatment leads to considerable improvement.31 However, stem cell transplantation depends on the availability of suitable donors and still carries significant risks. Studies performed on mice with acid ceramidase deficiency have shown the effectiveness of enzyme replacement therapy. Treatment of these mice with a single injection of human acid ceramidase-encoding lentivector resulted in enhanced growth, decreased ceramide levels and cellular infiltrations, and increased lifespan. The severity of the disease was greatly diminished.32 In theory, Farber disease could be amenable to treatment with enzyme substitution, similar to other sphingolipidoses.33
Boris Hugle, MD, MSc, is a staff rheumatologist and researcher at the German Center for Pediatric and Adolescent Rheumatology, Garmisch-Partenkirchen, Germany.
Laura Mueller, outreach coordinator for Plexcera Therapeutics LLC, New York, directs Plexcera’s outreach to parents and physicians to help spread awareness for Farber disease and SMA-PME.
Thierry Levade, MD, PhD, is professor of Biochemistry and Molecular Biology at the Toulouse-Rangueil Medical School and the principal investigator for the Sphingolipids, Cell Death and Tumor Progression group at the Toulouse Cancer Research Center (CRCT INSERM).
Acknowledgments
The authors would like to thank Ivan Galanin for his assistance in the preparation of this manuscript. The technical assistance of M.A. Berges, E. Le Trionnaire (Laboratoire de Biochimie Métabolique, CHU Toulouse), S. Carpentier and V. Garcia (INSERM UMR1037, Toulouse) is also acknowledged.
Authors’ note: If you have patients similar to the one described herein, please contact the authors at [email protected] to learn how to enroll them in a clinical trial.
References
- Park JH, Schuchman EH. Acid ceramidase and human disease. Biochim Biophys Acta. 2006;1758:2133–2138.
- Levade T, Sandhoff K, Fridovich-Keil JL, et al. Acid Ceramidase Deficiency: Farber Lipogranulomatosis. In Valle D, Beaudet AL, Vogelstein B, Kinzler KW, eds. Scriver’s Online Metabolic and Molecular Bases of Inherited Disease. http://dx.doi.org/10.1036/ommbid.173. Published January 2006. Accessed Nov. 17, 2013.
- Fiumara A, Nigro F, Pavone L, et al. Farber disease with prolonged survival. J Inherit Metab Dis. 1993;16:915–916.
- Moser H. Farber disease: Acid ceramidase deficiency and Farber lipogranulomatosis. In The Molecular and Genetic Basis of Neurologic and Psychiatric Disease. 4th edition. Edited by Rosenberg R. Philadelphia: Lippincott Williams & Wilkins; 2008:282–288.
- Sugita M, Dulaney JT, Moser HW. Ceramidase deficiency in Farber’s disease (lipogranulomatosis). Science. 1972;178:1100–1102.
- Antonarakis SE, Valle D, Moser HW, et al. Phenotypic variability in siblings with Farber disease. J Pediatr. 1984;104:406–409.
- Samuelsson K, Zetterstrom R. Ceramides in a patient with lipogranulomatosis (Farber’s disease) with chronic course. Scand J Clin Lab Invest. 1971;27:393–405.
- Burck U, Moser HW, Goebel HH, et al. A case of lipogranulomatosis Farber: Some clinical and ultrastructural aspects. Eur J Pediatr. 1985;143:203–208.
- Fujiwaki T, Hamanaka S, Tate S, et al. Tissue accumulation of sulfatide and GM3 ganglioside in a patient with variant Farber disease. Clin Chim Acta. 1995;234:23–36.
- Levade T, Moser HW, Fensom AH, et al. Neurodegenerative course in ceramidase deficiency (Farber disease) correlates with the residual lysosomal ceramide turnover in cultured living patient cells. J Neurol Sci. 1995;134:108–114.
- Nowaczyk MJ, Feigenbaum A, Silver MM, et al. Bone marrow involvement and obstructive jaundice in Farber lipogranulomatosis: Clinical and autopsy report of a new case. J Inherit Metab Dis. 1996;19:655–660.
- Schafer A, Harzer K, Kattner E, et al. [Disseminated lipogranulomatosis (Farber disease) with hydrops fetalis]. Pathologe. 1996;17:145–149.
- Kattner E, Schafer A, Harzer K. Hydrops fetalis: Manifestation in lysosomal storage diseases including Farber disease. Eur J Pediatr. 1997;156:292–295.
- Kim YJ, Park SJ, Park CK, et al. A case of Farber lipogranulomatosis. J Korean Med Sci. 1998;13:95–98.
- Yeager AM, Uhas KA, Coles CD, et al. Bone marrow transplantation for infantile ceramidase deficiency (Farber disease). Bone Marrow Transplant. 2000;26:357–363.
- Bar J, Linke T, Ferlinz K, Neumann U, et al. Molecular analysis of acid ceramidase deficiency in patients with Farber disease. Hum Mutat. 2001;17:199–209.
- Muramatsu T, Sakai N, Yanagihara I, et al. Mutation analysis of the acid ceramidase gene in Japanese patients with Farber disease. J Inherit Metab Dis. 2002;25:585–592.
- Salo MK, Karikoski R, Hallstrom M, et al. Farber disease diagnosed after liver transplantation. J Pediatr Gastroenterol Nutr. 2003;36:274–277.
- Devi AR, Gopikrishna M, Ratheesh R, et al. Farber lipogranulomatosis: Clinical and molecular genetic analysis reveals a novel mutation in an Indian family. J Hum Genet. 2006;51:811–814.
- Willis A, Vanhuse C, Newton KP, et al. Farber’s disease type IV presenting with cholestasis and neonatal liver failure: Report of two cases. Pediatr Dev Pathol. 2008;11:305–308.
- Ahmad A, Mazhar AU, Anwar M. Farber disease: A rare neurodegenerative disorder. J Coll Physicians Surg Pak. 2009;19:67–68.
- Sana C, Larbi E, Samir A, et al. Farber disease in a newborn. Pediatr Dermatol. 2009;26:44–46.
- Cvitanovic-Sojat L, Gjergja Juraski R, Sabourdy F, et al. Farber lipogranulomatosis type 1—late presentation and early death in a Croatian boy with a novel homozygous ASAH1 mutation. Eur J Paediatr Neurol. 2011;15:171–173.
- Chedrawi AK, Al-Hassnan ZN, Al-Muhaizea M, et al. Novel V97G ASAH1 mutation found in Farber disease patients: Unique appearance of the disease with an intermediate severity, and marked early involvement of central and peripheral nervous system. Brain Dev. 2012;34:400–404.
- Ekici B, Kurkcu D, Caliskan M. Farber disease: A clinical diagnosis. J Pediatr Neurosci. 2012;7:154–155.
- Muranjan M, Agarwal S, Lahiri K, et al. Novel biochemical abnormalities and genotype in Farber disease. Indian Pediatr. 2012;49:320–322.
- Alves MQ, Le Trionnaire E, Ribeiro I, et al. Molecular basis of acid ceramidase deficiency in a neonatal form of Farber disease: Identification of the first large deletion in ASAH1 gene. Mol Genet Metab. 2013;109:276–281.
- Jarisch A, Steward CG, Sorensen J, et al. Odontoid infiltration and spinal compression in Farber disease: Reversal by haematopoietic stem cell transplantation. Eur J Pediatr. July 2013.
- Kostik MM, Chikova IA, Avramenko VV, et al. Farber lipogranulomatosis with predominant joint involvement mimicking juvenile idiopathic arthritis. J Inherit Metab Dis. 2013;36:1079–1080.
- Moser H, Linke T, Fensom AH, et al. Acid ceramide deficiency: Farber lipogranulomatosis. In The Metabolic and Molecular Bases of Inherited Disease. 8th edition. Edited by Scriver CR. New York: McGraw-Hill. 2001;3573–3585.
- Ehlert K, Frosch M, Fehse N, et al. Farber disease: Clinical presentation, pathogenesis and a new approach to treatment. Pediatr Rheumatol Online J. 2007;5:15.
- Alayoubi AM, Wang JC, Au BC, et al. Systemic ceramide accumulation leads to severe and varied pathological consequences. EMBO Mol Med. 2013;5:827–842.
- Desnick RJ, Schuchman EH: Enzyme replacement therapy for lysosomal diseases: Lessons from 20 years of experience and remaining challenges. Annu Rev Genomics Hum Genet. 2012;13:307–335.