Medical, social & family history
The patient had been diagnosed with fibromyalgia 25 years earlier. She had hypercholesterolemia, hypertension, migraines, rhinitis and asthma. She was taking amitriptyline, aspirin, oxycodone/acetaminophen, pravastatin, sertraline, verapamil, carbamazepine and pantoprazole. She had a remote surgical history of appendectomy, cutaneous basal cell carcinoma and squamous cell carcinoma on her forehead. She also had a history of small meningioma. She did not drink alcohol or smoke. She was allergic to a sulfa antibiotic, which caused rash and laryngeal edema, and to tetracycline, which caused tongue swelling.
Her maternal and paternal families both originated in Czechoslovakia, and there was no known family history of periodic fever syndromes.
Review of systems
Our initial review of systems revealed fatigue, headache, photosensitivity, occasional dry eyes and burning mouth, and low back pain. She had no weight loss, and denied oral ulcers, cough, shortness of breath and chest pain. She also denied abdominal pain, diarrhea, nausea or vomiting.
Physical examination
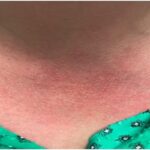
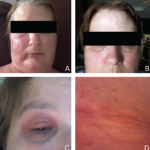
The patient’s vital signs were within normal limits, with a temperature of 97.8ºF. She had no rash, but she brought photographs showing facial erythema (see Figure 1, above). She had no palpable lymphadenopathy, oral ulcers, conjunctivitis or parotid gland enlargement. The patient had chest tenderness, but the cardiopulmonary examination revealed no murmurs, wheezing or rales. On abdominal examination, she had no tenderness, distention, rebound or hepatosplenomegaly. The musculoskeletal examination disclosed no tenderness, redness, warmth or swelling in her joints, and she had no signs of instability of the knees. Her muscle strength was normal, and she had no neurological deficits.
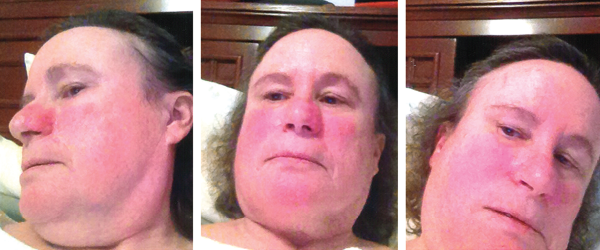
Figure 1: Erythema
Patchy erythema is seen on the cheeks, the bridge of nose, chin and neck.
Laboratory & radiographic evaluation
Complete blood counts, complete metabolic panels and urinalyses were essentially normal on multiple occasions, except for occasional, mild leukocytosis, thrombocytosis and eosinophilia. Her C-reactive protein (CRP) was 12.7 (normal <5 mg/L), and her erythrocyte sedimentation rate (ESR) and serum amyloid A (SAA) were normal.
Skin testing and radioallergosorbent (RAST) assays for both aeroallergens and food hypersensitivities, serum IgE and tryptase level all were negative or normal. Further laboratory tests, including stools for occult blood and lactoferrin, blood cultures, viral hepatitis, parvovirus, Lyme disease, Rickettsia, Babesia, Ehrlichia, Clostridium difficile and Aspergillus all came back negative.
Serologic markers were negative, including autoantibodies directed against nuclear antigens (ANA), extractable nuclear antigens (ENA), double-stranded DNA, neutrophil cytoplasmic antigens (ANCA), and cyclic citrullinated peptides, as well as rheumatoid factor and HLA-B27. Thyroid-stimulating hormone levels were within normal limits, and anti-thyroid peroxidase and thyroglobulin antibodies were negative.
Other tests, such as for creatinine kinase, aldolase, serum protein electrophoresis, uric acid and angiotensin-converting enzymes, were all negative or normal. Serum immunoglobulin quantitation, including IgG subclasses, IgE and IgD levels, was normal, except for IgA at 31 mg/dL (normal 81–463 mg/dL). Tissue transglutaminase IgA antibodies were negative. Genetic testing for celiac disease was negative for the HLA-DQ2/DQ8 haplotypes. Plasma cytokine (IL-1, IL-6 and TNFα) levels were normal.
A chest radiograph was unremarkable, but computerized tomography (CT) of the chest showed a small pericardial effusion. A subsequent echocardiogram was normal. Ultrasound examination of the abdomen was unremarkable; but CT scans of the abdomen and pelvis taken during attacks revealed mesenteric panniculitis (see Figure 2, opposite).
Esophagogastroduodenoscopy showed gastritis, and a colonoscopy was normal except for the presence of benign polyps.
A CT of the patient’s head revealed a left frontoparietal meningioma 0.9×0.7×1.1 cm, and a magnetic resonance angiography (MRA) of her brain was unremarkable.
A radiograph of the knees showed minimal degenerative disease in the left knee. A bone scan did not demonstrate evidence of malignancy, but showed a slight uptake in her carpi and feet.
Next-generation sequencing for periodic fever syndrome in a six-gene panel (MEFV, NLRP3, TNFRSF1A, MVK, NLRP12 and NOD2) was remarkable only for the nucleotide oligomerization domain, containing two (NOD2) variants: c.2798+158 C>T (IVS8+158) and c.2104C>T (R702W).
Differential Diagnoses
Infection: For the first few episodes of high fever and gastrointestinal (GI) symptoms, infection was a main concern. The patient had no recent travel history or animal exposure. Routine and specific tests for bacterial and viral etiology, as well as imaging studies, did not reveal any infection.
Allergic disorders: The patient’s medical history included intermittent asthma/bronchitis and rhinitis, and use of inhalers/nebulizers. She continued to have febrile episodes, with facial erythema, GI symptoms and myalgias. At this point, asthma and rhinitis focused us on allergic diseases, both common and uncommon, such as food-dependent, exercise-induced anaphylaxis and systemic mastocytosis (SM). However, skin testing and RAST assays, serum IgE, plasma IL-5 and tryptase levels all were negative or normal.
SM patients present with urticaria pigmentosa, lymphadenopathy, hepatosplenomegaly and/or patchy or diffusely increased tracer uptake on bone scanning.1 Serum tryptase level is elevated, and KIT mutation is detected in most patients with SM.2 Generally, fever and arthralgias are not seen in SM. The absence of the above manifestations argues against a diagnosis of SM in our case.
Carcinoid syndrome: This neuroendocrine tumor comprises symptoms of cutaneous flushing, diarrhea, cardiac valvular lesions, and bronchospasm related to excessive serotonin production.3 Symptoms of the syndrome are intermittent to progressive, and these symptoms usually do not occur until the primary tumor metastasizes. Clinical workup includes laboratory testing of 24-hour urinary excretion of the serotonin metabolites 5-hydroxyindoleacetic acid (5-HIAA) and plasma chromogranin A, and an imaging study. While our patient had some symptoms associated with carcinoid syndrome, her disease flares were intermittent and became less frequent and milder over time.4 In addition, fever is usually absent in carcinoid syndrome. Further, a CT scan of the chest, abdomen and pelvis, as well as endoscopies, did not disclose any signs of malignancy in the internal organs. In addition, her illness duration, relapsing and remitting course, and extensive workup made malignancy a less likely etiology.
Systemic autoimmune disease: After failure to substantiate a diagnosis of an allergic disorder, one would be concerned about a systemic autoimmune disease, such as systemic lupus erythematosus or vasculitis. However, there were no signs of any internal organ damage or cutaneous signs of vasculopathy (e.g., purpura, nodules, urticaria or ulcers). Moreover, complete serologic testing was negative. These data discount a diagnosis of a systemic autoimmune disease.
Inflammatory bowel disease (IBD) & celiac disease: Over the first two years, the patient continued to have episodic diffuse abdominal pain and explosive, non-bloody diarrhea, as well as diffuse myalgias, arthralgias affecting the knees and ankles and rash. Evaluation for IBD and celiac disease was completed. Esophagogastroduodenoscopy demonstrated gastritis, and a colonoscopy with tissue biopsies was unremarkable. CT of the abdomen/pelvis during acute attacks showed mesenteric signs of panniculitis (see Figure 2, right). A remote surgical history of appendectomy was noted, but there was no evidence of small bowel obstruction.
In IBD patients, lower extremity arthralgias/arthritis occurs in approximately 20%. Bloody diarrhea is common, and cutaneous findings more specific to IBD can be seen in 10% of patients, including erythema nodosum and pyoderma gangrenosum, whereas erythematous patches or plaques are astonishingly rare.5,6 Nevertheless, there was no endoscopic or tissue evidence of IBD in this patient. Dermatitis herpetiformis is more common in celiac disease, but was not seen in our patient.7 In addition to negative serology and duodenal pathology, genetic testing for celiac disease was negative, making a diagnosis of celiac disease unlikely.
Weber-Christian disease: Given the CT finding of abdominal panniculitis and fever, one would be concerned about Weber-Christian disease, though the disease concept is controversial. The disease is also known as relapsing febrile nodular nonsuppurative panniculitis. Subcutaneous nodules are common and prominent, but conspicuously absent from our patient.8 Generally, a CT finding of mesenteric panniculitis is a relatively common and nonspecific finding during evaluation of abdominal pain, and can be attributable to numerous causes.9 In our case, a repeat abdominal CT a month after resolution of her abdominal complaints showed disappearance of the radiographic finding.
Hereditary periodic fever syndromes: The patient continued to have episodic fever/chills, abdominal pain/diarrhea, and shortness of breath (asthmatic bronchitis), as well as rash and left knee pain/swelling. Within the first two years, three severe episodes prompted three separate hospitalizations. There was a small pericardial effusion on chest CT, but an echocardiogram was unremarkable. Her CRP was slightly elevated, and the ESR and SAA were normal. A detailed family history failed to identify evidence of a periodic fever syndrome. Regardless, a presumptive clinical diagnosis of familial Mediterranean fever (FMF) was made, and empiric oral colchicine 0.6 mg twice daily began. The patient initially felt better, but colchicine was eventually discontinued due to the lack of sustained efficacy. Subsequently, a targeted genetic analysis of MEFV did not demonstrate evidence of mutations associated with FMF.
Given the periodic nature of the disease course and lack of detectable autoantibodies in this case, we entertained a systemic autoinflammatory disease or periodic fever syndrome. Among these diseases are classic hereditary periodic fever syndromes.10
Typically, the FMF phenotype includes febrile episodes lasting less than 72 hours, lower extremity erysipeloid rash, and acute abdomen with more constipation than diarrhea.11 However, our patient did not present with the classic clinical characteristics, and complete sequencing of the MEFV gene failed to identify known mutations. Although she had a transient therapeutic response to colchicine, these data together argue against a diagnosis of FMF.
In cryopyrin-associated periodic syndrome (CAPS), also called NLRP3-associated autoinflammatory diseases, patients are often affected by generalized cold-induced urticaria, conjunctivitis and sensorineural hearing loss associated with NLRP3 mutations that may be revealed by gene sequencing.12-14
Tumor necrosis factor receptor-associated periodic syndrome (TRAPS) patients typically have abdominal pain, fevers lasting ~14 days, localized myalgias involving a group of muscles overlying eruptions, pleurisy, and periorbital edema, and TRAPS is associated with mutations of the TNFRSF1A gene.15
In hyper-IgD syndrome (HIDS) or mevalonate kinase (MVK) deficiency, disease onset occurs almost exclusively in children, and this disease shares similar clinical phenotype with other hereditary periodic fever syndromes. A more specific phenotype may consist of generalized lymphadenopathy, symmetric polyarthritis, and erythematous macules/papules. Genetic testing usually demonstrates MVK gene mutations.16
NOD2-associated diseases: The patient underwent a six-gene panel next-generation sequencing of her blood cells for periodic fever syndrome, and the molecular analysis was positive and heterozygous only for the NOD2 variants, IVS8+158 and R702W. Further laboratory study showed that blood CD4/CD8 T cell ratio was elevated, and B lymphocyte count was normal. Plasma cytokine levels were normal. Circulating vascular endothelial growth factor (VEGF) level was 604 pg/mL (normal <393 pg/mL), and intercellular adhesion molecule 1 (ICAM-1)/vascular cell adhesion molecule 1 (VCAM-1) was normal.
With identification of the NOD2 variants, we then focused our diagnosis on another subset of autoinflammatory diseases, which are associated with NOD2 variants.17 These diseases largely encompass Crohn’s disease, Blau syndrome and Yao syndrome.
Blau syndrome (BS) typically manifests as a triad of granulomatous dermatitis, uveitis and inflammatory arthritis. The disease primarily occurs in children as an autosomal dominant disorder, and adult-onset disease is extremely meager.18 Dermatitis commonly comprises erythematous papular scaly rash. Arthritis affects the wrist, knee, ankle and proximal interphalangeal joints (PIPs), leading to campytodactyly (flexion of PIPs) and rheumatoid arthritis-like and cystic articular changes.19 Both dermatitis and arthritis are nearly symmetric and persistent. Of note, fever and GI symptoms are unusual in the disease.19
Yao Syndrome (YAOS, OMIM617321)
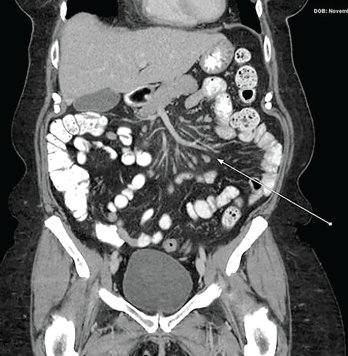
Figure 2: CT of the Abdomen & Pelvis with Contrast
The arrow shows findings of mesenteric panniculitis manifested as increase in density of the mesenteric fat surrounding the mesenteric vessels and increase in size and number of mesenteric lymph nodes.
YAOS, formerly designated as NOD2-associated autoinflammatory disease, is characterized by periodic fever, dermatitis, arthritis and swelling of the distal extremities, as well as GI and sicca-like symptoms.6,20-22 Patients generally have flu-like symptoms, followed by episodic fever and erythematous patches or plaques with pathological finding of spongiotic dermatitis mostly. An episodic flare may last a few days to weeks with an asymptomatic interval of several weeks or months.
GI symptoms are common, including recurrent abdominal pain, cramping, bloating and non-bloody diarrhea, and the symptoms may last several days to weeks. There is no endoscopic, radiological or histopathological evidence of IBD. Oligo- or polyarthritis is common, and occurs periodically without joint erosions, destruction or deformities; joints appear normal when the disease is quiescent. Approximately 25% patients develop intermittent, unilateral or bilateral, distal lower extremity swelling involving the ankles and feet.23 In addition, patients can have oral ulcers and chest pain from pericarditis or/and pleuritis.
The proposed diagnostic criteria for YAOS comprise the characteristic phenotype, specific NOD2 variants and rigorous exclusion of overlapping disease entities.22 The NOD2 variant IVS8+158 is detected in almost all YAOS patients, with concurrent R702W in up to 30%; other, rarer NOD2 variants can be identified in some patients.21
YAOS has been predominantly reported in white adults, with a female to male ratio of 2:1. The mean age of onset is 30 years. The disease occurs sporadically in a majority of patients, and a positive family history is seen in a minority. Although it is a multi-organ disease, YAOS rarely affects solid internal organs.
In addition to the characteristic clinical phenotype, these patients can have episodic asthma/shortness of breath and fibromyalgia/chronic pain syndrome as relatively common comorbidities.22 All in all, the clinical phenotype and genotype in our case indicated YAOS.
Patient’s Course
The therapeutic strategy for YAOS depends on the frequency and severity of the disease flares. Treatment remains empiric and involves intermittent short courses (i.e., several days) of glucocorticoids, sulfasalazine and/or IL-1 inhibitors.22,24 The patient was followed at our rheumatology clinic every six months, during which time she might have one or two mild flares, including fever, rash and abdominal pain. Her flares were managed with short courses of prednisone and symptomatic treatment. Sulfasalazine was not given due to her prior allergy, and the use of any anti-IL-1 biologics was deferred because of more recent mild disease.
Discussion
Systemic autoinflammatory diseases (SAIDs) have been categorized as a genetically heterogeneous group of rheumatic and inflammatory diseases over the past 20 years.25 This group of rheumatic disorders has undergone a rapid expansion, including, but not limited to, the hereditary periodic fever syndromes.26 These diseases are distinct from autoimmune diseases, such as systemic lupus erythematosus, rheumatoid arthritis and systemic vasculitis. The former group of disorders is often recurrent and possesses a constellation of fever, rash, arthralgias, abdominal and/or chest pain. More importantly, these diseases are usually devoid of detectable autoantibodies and are often associated with genetic markers/mutations.
SAIDs often share similar clinical phenotypes with one another, and they can mimic infectious, allergic, immunologic and autoimmune disorders, particularly in the early stage of disease, as in our case.27 Therefore, a broad differential diagnosis should be considered, including the new group of autoinflammatory disorders.
Classic SAIDs are often inherited in either autosomal recessive (FMF and HIDS) or dominant (CAPS and TRAPS) pattern, and thus, early onset of disease is common along with a positive family history, although late onset of these diseases can occur and present in an atypical or sporadic way. In the classic periodic fever syndromes, based upon our clinical experience and study, FMF, CAPS and TRAPs are clinically encountered in a decreasing order in the adult patient population, and adult onset of HIDS is extremely rare.28
The spectrum of NOD2-associated diseases has undergone an expansion over the past 10 years.29 NOD2 is a cytosolic innate immune sensor, which recognizes and responds to peptidoglycan contained within the cell wall of bacteria; it contributes to immune homeostasis.17
Crohn’s disease is associated with the three common loss-of-function NOD2 mutations, 1007fs, G908R and R702W, in up to 40% of cases, although the disease is also associated with variants in other multiple genes.30,31
BS is linked to the high-penetrant NOD2 variants in the central region. NOD and these NOD2 sequence variants include, but are not limited to, R334W, R334Q, E383K, E383G, G464W, L469F, W490L, M513R, R587C, T605N, H496L, M513T, C495Y and N670K, with R334W and R334Q being the most frequent variants.32 These variants represent gain of function.33
Since our initial description of the disease in 2011, YAOS has been reported in the U.S., Europe and Asia.6,34-37 To date, most cases have been reported in adulthood and are sporadic, without a clear pattern of inheritance. Disease onset can occur in children.
The pathogenesis of YAOS remains elusive. Our study has shown that NOD2 expression at transcriptional levels and pathway activation is abnormal in the disease. Specific NOD2 genotypes result in distinct NOD2 expression and cytokine profiles.38 In this disease, serum immunoglobulin levels can be low, and plasma levels of cytokines are normal as in this case.38,39
Other serum factors and adaptive immune responses have not been studied, although autoinflammatory disease is conceptually defined to mainly affect innate immune response. YAOS is considered to be polygenic; the interplay between the deficient NOD2 function, other innate immune sensor mutations and the environment is postulated to contribute to the disease. To prove this hypothesis, further study will be needed.
The disease is not uncommon; the incidence has been estimated at 1–10 cases per 100,000 people in the adult population.22
In conclusion, several important points are raised from this complex case analysis.
- In evaluating patients with episodic symptoms/manifestations, physicians need to be alert for autoinflammatory diseases;
- Although SAIDs mostly occur in children, such disorders can present in adults;
- Physicians need to recognize relatively new SAIDs beyond the hereditary periodic fever syndromes; and
- Proper application of the periodic fever syndrome gene panel is helpful for early diagnosis, and enables reduction of unnecessary and repetitive testing.
 Peter Gorevic, MD, is a rheumatologist, professor of medicine, and former chair of the Department of Rheumatology, Icahn School of Medicine, Mount Sinai Hospital, N.Y.
Peter Gorevic, MD, is a rheumatologist, professor of medicine, and former chair of the Department of Rheumatology, Icahn School of Medicine, Mount Sinai Hospital, N.Y.
 Qingping Yao, MD, PhD, is a rheumatologist, Zhang Family Endowed Chair in Rheumatology, professor in the Department of Medicine, chief of the Division of Rheumatology, Allergy and Immunology, and director of the Center of Autoinflammatory Diseases, Stony Brook University Renaissance School of Medicine, N.Y.
Qingping Yao, MD, PhD, is a rheumatologist, Zhang Family Endowed Chair in Rheumatology, professor in the Department of Medicine, chief of the Division of Rheumatology, Allergy and Immunology, and director of the Center of Autoinflammatory Diseases, Stony Brook University Renaissance School of Medicine, N.Y.
Acknowledgments
The authors would like to thank Alan Kaell, MD, clinical professor of medicine at Stony Brook University, and Clara Abraham, MD, professor of medicine, Section of Digestive Diseases at Yale University, New Haven, Conn., for their kind review of the manuscript and comments. We are also thankful to Matthew Barish, MD, FACR, associate professor of clinical radiology, vice chair, Department of Radiology at Stony Brook University for his review of the CT images, and Ms. Lyn Hastings, communication specialist, Department of Medicine at Stony Brook University, for making the composite photograph.
References
- Gotlib J, Kluin-Nelemans HC, George TI, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med. 2016 Jun 30;374:2530–2541.
- Cruse G, Metcalfe DD, Olivera A. Functional deregulation of KIT: Link to mast cell proliferative diseases and other neoplasms. Immunol Allergy Clin North Am. 2014 May;34(2):219–237.
- Maton PN. The carcinoid syndrome. JAMA. 1988 Sep 16;260(11):1602–1605.
- Rastogi V, Singh D, Mazza JJ, et al. Flushing disorders associated with gastrointestinal symptoms: Part 1, neuroendocrine tumors, mast cell disorders and hyperbasophila. Clin Med Res. 2018 Jun;16(1–2):16–28.
- Farhi D, Cosnes J, Zizi N, et al. Significance of erythema nodosum and pyoderma gangrenosum in inflammatory bowel diseases: A cohort study of 2,402 patients. Medicine (Baltimore). 2008 Sep;87(5):281–293.
- Yao Q, Su LC, Tomecki KJ, et al. Dermatitis as a characteristic phenotype of a new autoinflammatory disease associated with NOD2 mutations. J Am Acad Dermatol. 2013 Apr;68(4):624–631.
- Rodrigues F, Bachmeyer C. Coeliac disease and dermatitis herpetiformis. Lancet. 2018 Sep 15;392(10151):916.
- White JW Jr., Winkelmann RK. Weber-Christian panniculitis: A review of 30 cases with this diagnosis. J Am Acad Dermatol. 1998 Jul;39(1):56–62.
- van Breda Vriesman AC, Schuttevaer HM, Coerkamp EG, et al. Mesenteric panniculitis: US and CT features. Eur Radiol. 2004 Dec;14(12):2242–2248.
- Yao Q, Furst DE. Autoinflammatory diseases: An update of clinical and genetic aspects. Rheumatology (Oxford). 2008 Jul;47:946–951.
- Mor A, Gal R, Livneh A. Abdominal and digestive system associations of familial Mediterranean fever. Am J Gastroenterol. 2003 Dec;98(12):2594–2604.
- Aksentijevich I, Putnam CD, Remmers EF, et al. The clinical continuum of cryopyrinopathies: Novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007 Apr;56(4):1273–1285.
- Georgin-Lavialle S, Fayand A, Rodrigues F, et al. Autoinflammatory diseases: State of the art. Presse Med. 2019 Feb;48(1 Pt 2):e25–e48.
- Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin 1 TRAP) in patients with cryopyrin-associated periodic syndromes: Results from two sequential placebo-controlled studies. Arthritis Rheum. 2008;58(8):2443–2452.
- Yao Q, Englund KA, Hayden SP, Tomecki KJ. Tumor necrosis factor receptor associated periodic fever syndrome with photographic evidence of various skin disease and unusual phenotypes: Case report and literature review. Semin Arthritis Rheum. 2012 Feb;41(4):611–617.
- Drenth JP, Cuisset L, Grateau G, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet. 1999 Jun;22(2):178–181.
- Yao Q. Nucleotide-binding oligomerization domain containing 2: Structure, function and diseases. Semin Arthritis Rheum. 2013 Aug;43(1):125–130
- Yao Q, Piliang M, Nicolacakis K, et al. Granulomatous pneumonitis associated with adult-onset Blau-like syndrome. Am J Respir Crit Care Med. 2012 Sep 1;186(5):465–466.
- Rose CD, Pans S, Casteels I, et al. Blau syndrome: Cross-sectional data from a multicentre study of clinical, radiological and functional outcomes. Rheumatology (Oxford). 2015 Jun;54(6):1008–1016.
- Yao Q, Zhou L, Cusumano P, et al. A new category of autoinflammatory disease associated with NOD2 gene mutations. Arthritis Res Ther. 2011;13(5):R148.
- Yao Q, Shen M, McDonald C, et al. NOD2-associated autoinflammatory disease: A large cohort study. Rheumatology (Oxford). 2015 Oct;54(10):1904–1912.
- Yao Q, Shen B. A systematic analysis of treatment and outcomes of NOD2-associated autoinflammatory disease. Am J Med. 2017 Mar;130(3):365.e13–e18.
- Yao Q, Schils J. Distal lower extremity swelling as a prominent phenotype of NOD2-associated autoinflammatory disease. Rheumatology (Oxford). 2013 Nov;52(11):2095–2097.
- Yao Q. Research letter: Effectiveness of canakinumab for the treatment of Yao syndrome patients. J Am Acad Dermatol. 2019 Sep 18;S0190-9622(19)32757-4.
- De Benedetti F, Gattorno M, Anton J, et al. Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N Engl J Med. 2018 May 17;378(20):1908–1919.
- Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: A clinical perspective. Cell. 2010 Mar 19;140(6):784–790.
- Gattorno M, Hofer M, Federici S, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis. 2019 Aug;78(8):1025–1032.
- Yao Q, Lacbawan F, Li J. Adult autoinflammatory disease frequency and our diagnostic experience in an adult autoinflammatory clinic. Semin Arthritis Rheum. 2016 Apr;45(5):633–637.
- Yao Q, Li E, Shen B. Autoinflammatory disease with focus on NOD2-associated disease in the era of genomic medicine. Autoimmunity. 2019 Mar;52(2):48–56.
- Lesage S, Zouali H, Cezard JP, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002 Apr;70(4):845–857.
- Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012 Nov 1;491(7422):119–124.
- Wu D, Shen M. Two Chinese pedigrees of Blau syndrome with thirteen affected members. Clin Rheumatol. 2018 Jan;37(1):265–270.
- Rose CD, Martin TM, Wouters CH. Blau syndrome revisited. Curr Opin Rheumatol. 2011 Sep;23(5):411–418.
- Estephan M, Yao Q, Springer J. Case of NOD2-associated autoinflammatory disease successfully treated with sulfasalazine. J Clin Rheumatol. 2017 Jan;23(1):58–59.
- Dziedzic M, Marjanska A, Babol-Pokora K, et al. Co-existence of Blau syndrome and NAID? Diagnostic challenges associated with presence of multiple pathogenic variants in NOD2 gene: A case report. Pediatr Rheumatol Online J. 2017 Jul 27;15(1):57.
- Trueb B, Zhuang L, Keller I, et al. Coincidence of NOD2-associated autoinflammatory disease (Yao syndrome) and HCV infection with fatal consequences: Interaction between genes and environment. J Clin Rheumatol. 2018 Dec 28. [Epub ahead of print].
- Yang X, Wu D, Li J, et al. A Chinese case series of Yao syndrome and literature review. Clin Rheumatol. 2018 Dec;37(12):3449–3454.
- McDonald C, Shen M, Johnson EE, et al. Alterations in nucleotide-binding oligomerization domain-2 expression, pathway activation, and cytokine production in Yao syndrome. Autoimmunity. 2018 Mar;51(2):53–61.
- Yao Q, Shen M, Fernandez J. NOD2-associated autoinflammatory disease and immune deficiency. J Allergy Clin Immunol Pract. 2016 Jul–Aug;4(4):780–782.