Immune-osteoblast Interactions
Although osteoclasts have been a major focus of attention in the study of inflammatory bone loss, surprisingly little is known about the activity of osteoblasts in this setting. Nevertheless, because bone metabolism depends on the balance between bone-resorbing osteoclasts and bone-forming osteoblasts, reduced bone formation likely contributes to bone loss in patients with arthritis. The mechanism behind this interaction is not fully defined, although TNF and other factors may repress bone formation, at least in part through the induction of dickkopf (Dkk)-1 and sclerostin, both inhibitors of the Wnt signaling, which is a central pathway for bone formation.13 Whereas Dkk-1 and sclerostin may act to suppress bone responses in diseases like rheumatoid arthritis (RA), impaired expression of these molecules also may be linked to aberrant bone formation, as found in spondyloarthritis (SpA). In addition to the effects of Wnt signaling, bone morphogenic proteins and transforming growth factor-β may induce new bone formation in arthritis.14 (See Figure 2, p. 37.)
Bone Erosion in RA
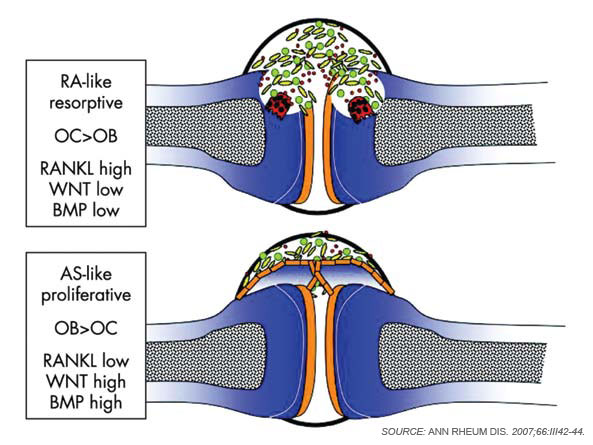
Inflammation is the primary trigger for the process of local bone erosions in RA, consistent with the prominence of inflammatory cell infiltrates in the tissue of patients with this disease (see Figure 3, at right). Bromley and Woolley and Gravallese and colleagues provided the first detailed descriptions of the location and activity of osteoclasts in the RA joint.15,16 These investigators demonstrated the expression of osteoclasts at the interface between advancing pannus tissue and the bone surface. Later, several groups demonstrated the essential role of osteoclasts in triggering inflammatory bone erosions in experimental disease models by blocking essential molecules for osteoclastogenesis or studying osteoclast-deficient mice.2,4